
Cardioprotective Effects of Palmitoleic Acid (C16:1n7) in a Mouse Model of Catecholamine-Induced Cardiac Damage Are Mediated by PPAR Activation

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
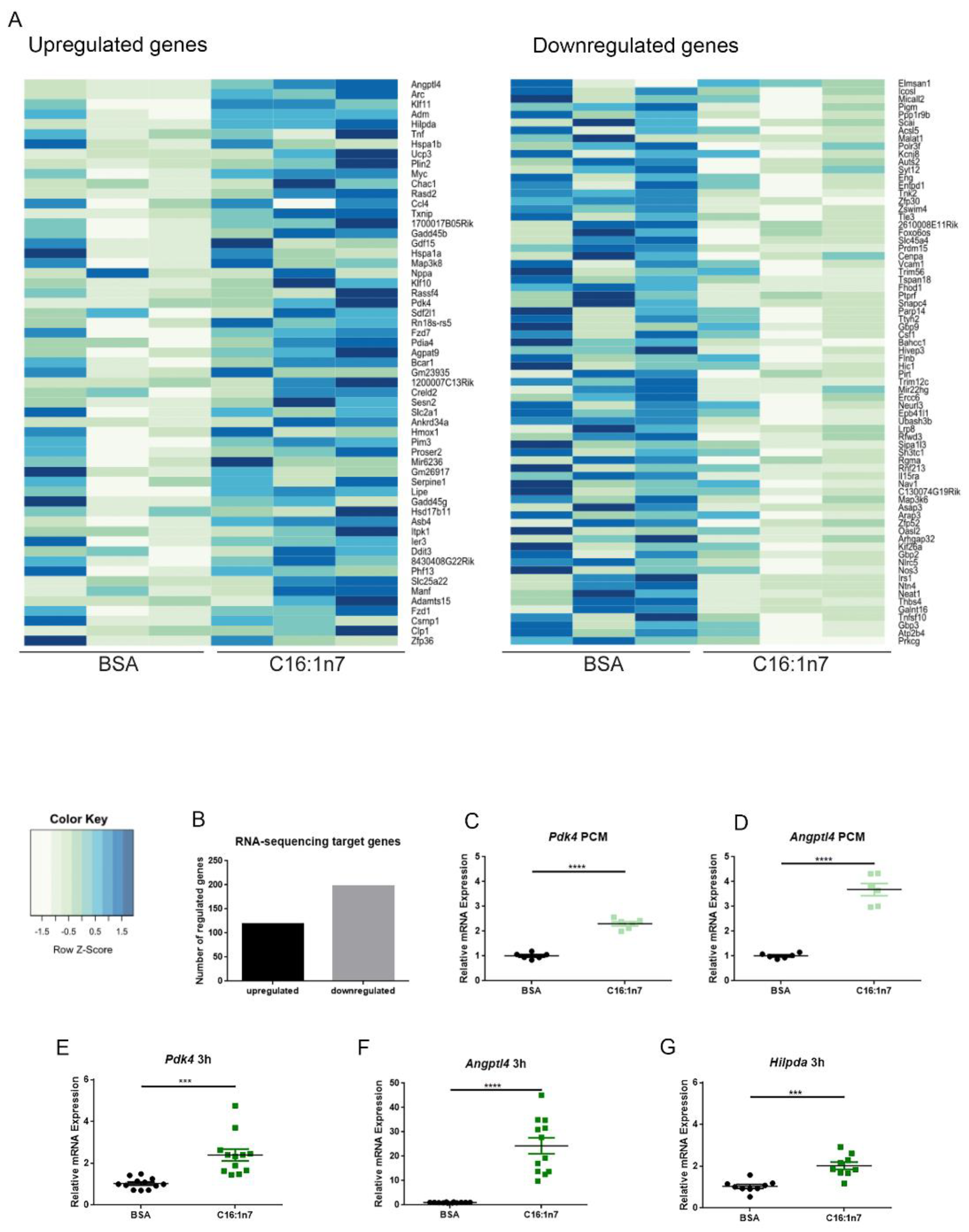
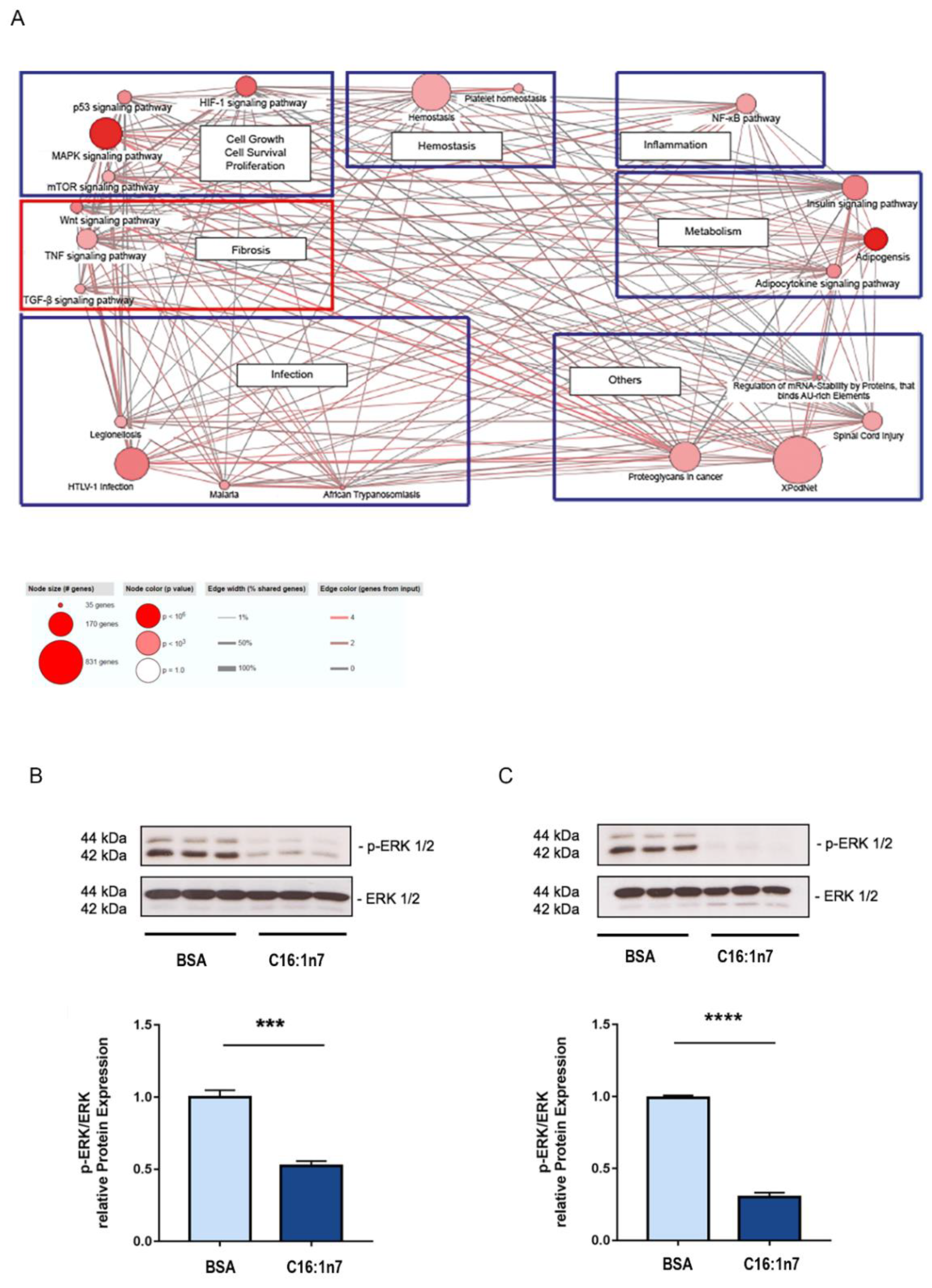
2.1. C16:1n7 Induces PPARα/δ-Specific Gene Expression Profile in the Primary Adult Murine Cardiomyocytes (PCMs)
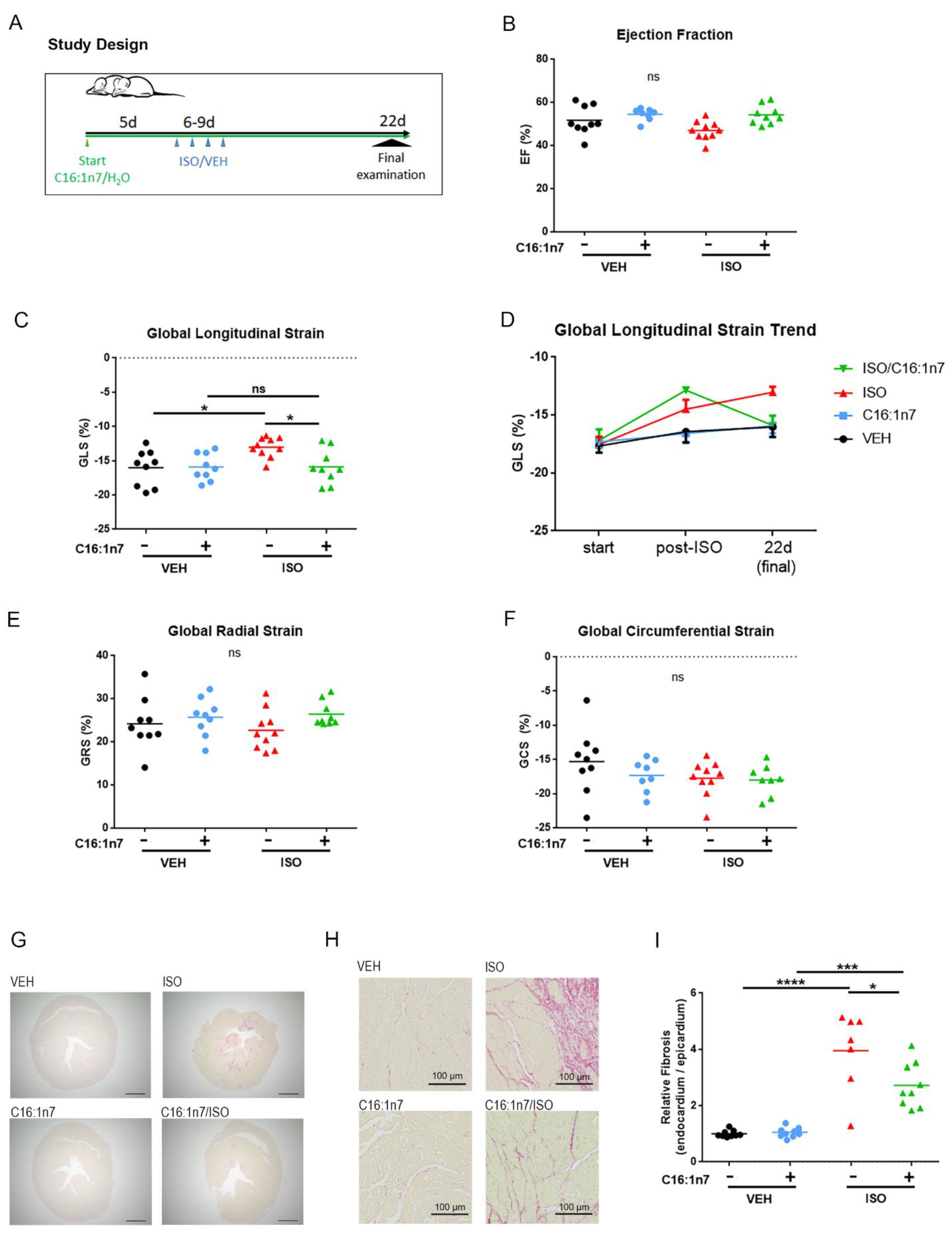
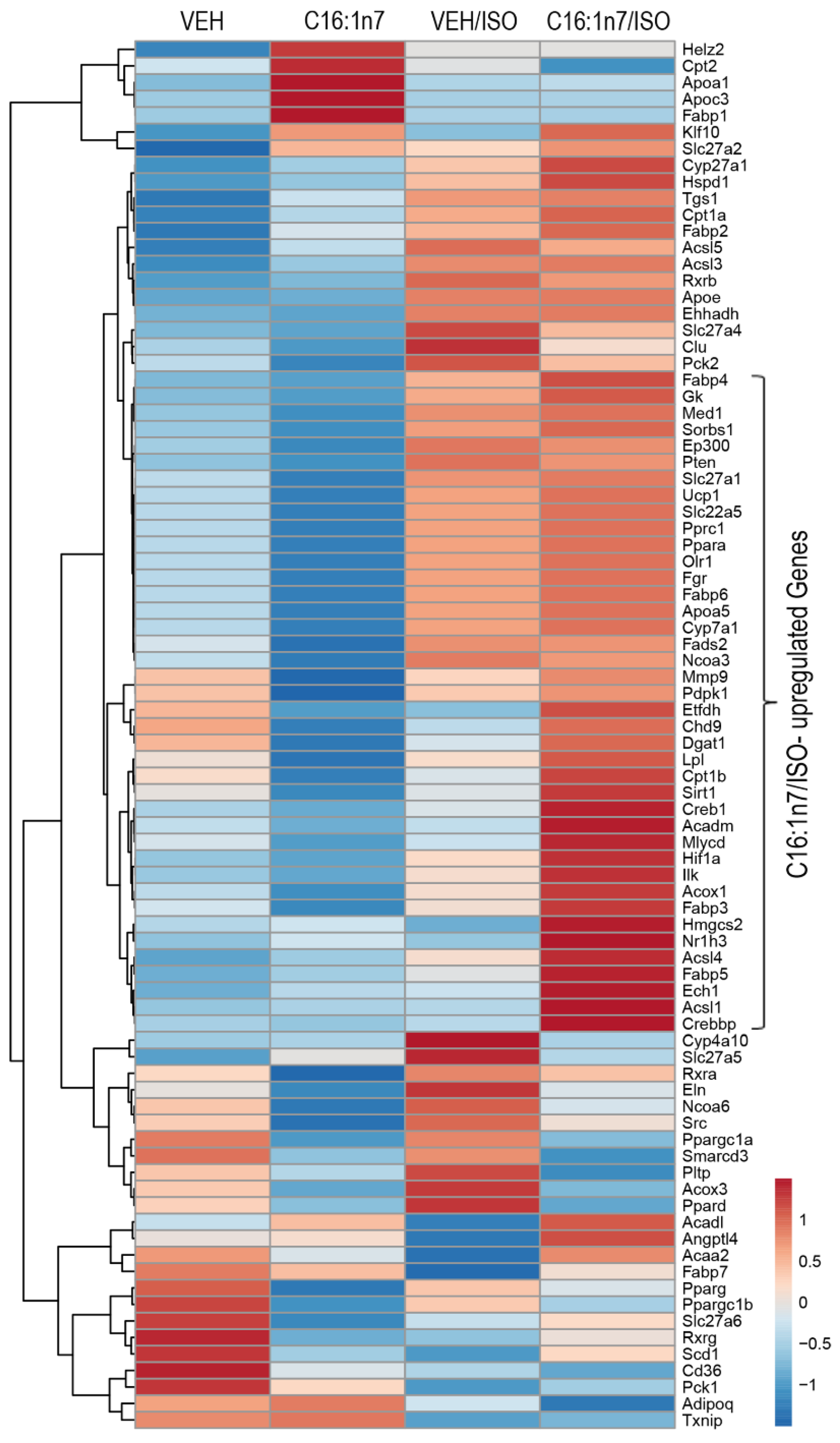
2.2. C16:1n7 Displays Anti-Fibrotic and Anti-Inflammatory Action in the Model of ISO-Induced Cardiac Damage in Mice
3. Discussion
4. Materials and Methods
4.1. Isolation of Adult Murine Primary Cardiomyocytes (PCMs)
4.2. Animal Experiments
4.3. Cell Culture Experiments with HL-1 Cardiomyocytes and PCMs
4.4. PCMs-RNA Sequencing and Pathway Analysis
4.5. Pathway Analysis Using ConsensusPath DB (CPDB)
4.6. Western Blot Analysis
4.7. Gene Expression Analysis
4.8. PPAR-Specific Gene Expression Analysis
4.9. Histology
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauersachs, J. Heart failure drug treatment: the fantastic four. Eur. Heart J. 2021, 42, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M.; Rolett, E.L. Heart failure: when form fails to follow function. Eur. Heart J. 2016, 37, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Florea, V.G.; Cohn, J.N. The autonomic nervous system and heart failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Beyhoff, N.; Brix, S.; Betz, I.R.; Klopfleisch, R.; Foryst-Ludwig, A.; Krannich, A.; Stawowy, P.; Knebel, F.; Grune, J.; Kintscher, U. Application of Speckle-Tracking Echocardiography in an Experimental Model of Isolated Subendocardial Damage. J. Am. Soc. Echocardiogr. 2017, 30, 1239–1250.e1232. [Google Scholar] [CrossRef]
- Beyhoff, N.; Lohr, D.; Foryst-Ludwig, A.; Klopfleisch, R.; Brix, S.; Grune, J.; Thiele, A.; Erfinanda, L.; Tabuchi, A.; Kuebler, W.M.; et al. Characterization of Myocardial Microstructure and Function in an Experimental Model of Isolated Subendocardial Damage. Hypertension 2019, 74, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Ritter, D.; Goeritzer, M.; Thiele, A.; Blumrich, A.; Beyhoff, N.; Luettges, K.; Smeir, E.; Kasch, J.; Grune, J.; Muller, O.J.; et al. Liver X Receptor Agonist AZ876 Induces Beneficial Endogenous Cardiac Lipid Reprogramming and Protects Against Isoproterenol-Induced Cardiac Damage. J. Am. Heart Assoc. 2021, 10, e019473. [Google Scholar] [CrossRef]
- Thiele, A.; Luettges, K.; Ritter, D.; Beyhoff, N.; Smeir, E.; Grune, J.; Steinhoff, J.S.; Schupp, M.; Klopfleisch, R.; Rothe, M.; et al. Pharmacological inhibition of adipose tissue Adipose Triglyceride Lipase (ATGL) by Atglistatin prevents catecholamine-induced myocardial damage. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Rawlins, J.; Bhan, A.; Sharma, S. Left ventricular hypertrophy in athletes. Eur. J. Echocardiogr. 2009, 10, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, J.E.; Vork, M.M.; Roemen, T.H.; de Jong, Y.F.; Cleutjens, J.P.; van der Vusse, G.J.; van Bilsen, M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J. Lipid Res. 1997, 38, 1384–1394. [Google Scholar] [CrossRef]
- Paumen, M.B.; Ishida, Y.; Muramatsu, M.; Yamamoto, M.; Honjo, T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J. Biol. Chem 1997, 272, 3324–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahey, R.; Wang, X.; Carley, A.N.; Lewandowski, E.D. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation 2014, 130, 1790–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, J.; Arita, M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J. Cardiol. 2016, 67, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, C.A.; Magida, J.A.; Harrison, B.C.; Wall, C.E.; Marr, T.G.; Secor, S.M.; Leinwand, L.A. Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science 2011, 334, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Foryst-Ludwig, A.; Kreissl, M.C.; Benz, V.; Brix, S.; Smeir, E.; Ban, Z.; Januszewicz, E.; Salatzki, J.; Grune, J.; Schwanstecher, A.K.; et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J. Biol. Chem. 2015, 290, 23603–23615. [Google Scholar] [CrossRef] [Green Version]
- de Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schunemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Zhou, Q.; Kwame Amakye, W.; Su, Y.; Zhang, Z. Biomarkers of dairy fat intake and risk of cardiovascular disease: A systematic review and meta analysis of prospective studies. Crit. Rev. Food Sci. Nutr. 2018, 58, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.; Sala-Vila, A.; Plana, N.; Girona, J.; Vallve, J.C.; Ibarretxe, D.; Ros, E.; Ferre, R.; Heras, M.; Masana, L. Serum palmitoleate acts as a lipokine in subjects at high cardiometabolic risk. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Frigolet, M.E.; Gutierrez-Aguilar, R. The Role of the Novel Lipokine Palmitoleic Acid in Health and Disease. Adv. Nutr. 2017, 8, 173S–181S. [Google Scholar] [CrossRef]
- Djousse, L.; Weir, N.L.; Hanson, N.Q.; Tsai, M.Y.; Gaziano, J.M. Plasma phospholipid concentration of cis-palmitoleic acid and risk of heart failure. Circ. Heart Fail. 2012, 5, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djousse, L.; Matthan, N.R.; Lichtenstein, A.H.; Gaziano, J.M. Red blood cell membrane concentration of cis-palmitoleic and cis-vaccenic acids and risk of coronary heart disease. Am. J. Cardiol. 2012, 110, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleber, M.E.; Delgado, G.E.; Lorkowski, S.; Marz, W.; von Schacky, C. Trans-fatty acids and mortality in patients referred for coronary angiography: the Ludwigshafen Risk and Cardiovascular Health Study. Eur. Heart J. 2016, 37, 1072–1078. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Zhao, L.; Yu, H.; Ma, X.; Bao, Y.; Rajani, C.; Loo, L.W.; Shvetsov, Y.B.; Yu, H.; Chen, T.; et al. Circulating Unsaturated Fatty Acids Delineate the Metabolic Status of Obese Individuals. EBioMedicine 2015, 2, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Mozaffarian, D.; Cao, H.; King, I.B.; Lemaitre, R.N.; Song, X.; Siscovick, D.S.; Hotamisligil, G.S. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am. J. Clin. Nutr. 2010, 92, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Kantartzis, K.; Celebi, N.; Staiger, H.; Machann, J.; Schick, F.; Cegan, A.; Elcnerova, M.; Schleicher, E.; Fritsche, A.; et al. Circulating palmitoleate strongly and independently predicts insulin sensitivity in humans. Diabetes Care 2010, 33, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Bolsoni-Lopes, A.; Festuccia, W.T.; Farias, T.S.; Chimin, P.; Torres-Leal, F.L.; Derogis, P.B.; de Andrade, P.B.; Miyamoto, S.; Lima, F.B.; Curi, R.; et al. Palmitoleic acid (n-7) increases white adipocyte lipolysis and lipase content in a PPARalpha-dependent manner. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1093–E1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salatzki, J.; Foryst-Ludwig, A.; Bentele, K.; Blumrich, A.; Smeir, E.; Ban, Z.; Brix, S.; Grune, J.; Beyhoff, N.; Klopfleisch, R.; et al. Adipose tissue ATGL modifies the cardiac lipidome in pressure-overload-induced left ventricular failure. PLoS Genet. 2018, 14, e1007171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foryst-Ludwig, A.; Kreissl, M.C.; Sprang, C.; Thalke, B.; Bohm, C.; Benz, V.; Gurgen, D.; Dragun, D.; Schubert, C.; Mai, K.; et al. Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H115-122. [Google Scholar] [CrossRef] [Green Version]
- Meng, R.; Pei, Z.; Zhang, A.; Zhou, Y.; Cai, X.; Chen, B.; Liu, G.; Mai, W.; Wei, J.; Dong, Y. AMPK activation enhances PPARalpha activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch. Biochem. Biophys. 2011, 511, 1–7. [Google Scholar] [CrossRef]
- Huang, Q.; Huang, J.; Zeng, Z.; Luo, J.; Liu, P.; Chen, S.; Liu, B.; Pan, X.; Zang, L.; Zhou, S. Effects of ERK1/2/PPARalpha/SCAD signal pathways on cardiomyocyte hypertrophy induced by insulin-like growth factor 1 and phenylephrine. Life Sci. 2015, 124, 41–49. [Google Scholar] [CrossRef]
- Liu, T.J.; Lai, H.C.; Ting, C.T.; Wang, P.H. Bidirectional regulation of upstream IGF-I/insulin receptor signaling and downstream FOXO1 in cardiomyocytes. J. Endocrinol. 2007, 192, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokharel, Y.; Sun, W.; Polfus, L.M.; Folsom, A.R.; Heiss, G.; Sharrett, A.R.; Boerwinkle, E.; Ballantyne, C.M.; Hoogeveen, R.C. Lipoprotein associated phospholipase A2 activity, apolipoprotein C3 loss-of-function variants and cardiovascular disease: The Atherosclerosis Risk In Communities Study. Atherosclerosis 2015, 241, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Gisonno, R.A.; Masson, T.; Ramella, N.A.; Barrera, E.E.; Romanowski, V.; Tricerri, M.A. Evolutionary and structural constraints influencing apolipoprotein A-I amyloid behavior. Proteins 2021. [Google Scholar] [CrossRef]
- Marozzi, M.; Parnigoni, A.; Negri, A.; Viola, M.; Vigetti, D.; Passi, A.; Karousou, E.; Rizzi, F. Inflammation, Extracellular Matrix Remodeling, and Proteostasis in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8102. [Google Scholar] [CrossRef]
- Francis, G.A.; Annicotte, J.S.; Auwerx, J. PPAR-alpha effects on the heart and other vascular tissues. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1-9. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Engel, F.B. PPARbeta/delta: Linking Metabolism to Regeneration. Int. J. Mol. Sci. 2018, 19, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffar, T.; Berube-Simard, F.A.; Bousette, N. Cardiomyocyte lipotoxicity is mediated by Il-6 and causes down-regulation of PPARs. Biochem. Biophys. Res. Commun. 2015, 459, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Oligino, E.; Rader, D.J.; Saghatelian, A.; Plutzky, J. VLDL hydrolysis by hepatic lipase regulates PPARdelta transcriptional responses. PLoS ONE 2011, 6, e21209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Muoio, D.M.; Way, J.M.; Tanner, C.J.; Winegar, D.A.; Kliewer, S.A.; Houmard, J.A.; Kraus, W.E.; Dohm, G.L. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002, 51, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef]
- Li, L.; Fang, H.; Yu, Y.H.; Liu, S.X.; Yang, Z.Q. Liquiritigenin attenuates isoprenalineinduced myocardial fibrosis in mice through the TGFbeta1/Smad2 and AKT/ERK signaling pathways. Mol. Med. Rep. 2021, 24. [Google Scholar] [CrossRef]
- Lago, R.M.; Singh, P.P.; Nesto, R.W. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: a meta-analysis of randomised clinical trials. Lancet 2007, 370, 1129–1136. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.; Guthof, T.; Pieske, B.M.; Heinzel, F.R.; Hohendanner, F. Isolation of Atrial Cardiomyocytes from a Rat Model of Metabolic Syndrome-related Heart Failure with Preserved Ejection Fraction. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J. Mol. Cell Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VEH | C16:1n7 | ISO | C16:1n7/ISO | |
|---|---|---|---|---|
| IVSd (mm) | 0.6613 ± 0.048 | 0.6488 ± 0.068 | 0.6817 ± 0.095 | 0.6191 ± 0.083 |
| PWd (mm) | 0.5818 ± 0.073 | 0.5626 ± 0.063 | 0.5639 ± 0.061 | 0.5567 ± 0.055 |
| LVIDd (mm) | 3.717 ± 0.202 | 3.602 ± 0.207 | 3.882 ± 0.174 | 3.890 ± 0.209 * |
| FS (%) | 26.12 ± 5.92 | 26.67 ± 5.89 | 24.85 ± 3.87 | 26.26 ± 2.33 |
| LVM (mg) | 74.65 ± 10.9 | 72.60 ± 5.46 | 80.92 ± 13.11 | 81.88 ± 7.51 |
| HW (mg) | 113.8 ± 5.95 | 113.7 ± 10.1 | 122.6 ± 8.63 | 121.3 ± 5.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betz, I.R.; Qaiyumi, S.J.; Goeritzer, M.; Thiele, A.; Brix, S.; Beyhoff, N.; Grune, J.; Klopfleisch, R.; Greulich, F.; Uhlenhaut, N.H.; et al. Cardioprotective Effects of Palmitoleic Acid (C16:1n7) in a Mouse Model of Catecholamine-Induced Cardiac Damage Are Mediated by PPAR Activation. Int. J. Mol. Sci. 2021, 22, 12695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312695
Betz IR, Qaiyumi SJ, Goeritzer M, Thiele A, Brix S, Beyhoff N, Grune J, Klopfleisch R, Greulich F, Uhlenhaut NH, et al. Cardioprotective Effects of Palmitoleic Acid (C16:1n7) in a Mouse Model of Catecholamine-Induced Cardiac Damage Are Mediated by PPAR Activation. International Journal of Molecular Sciences. 2021; 22(23):12695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312695
Chicago/Turabian StyleBetz, Iris Rosa, Sarah Julia Qaiyumi, Madeleine Goeritzer, Arne Thiele, Sarah Brix, Niklas Beyhoff, Jana Grune, Robert Klopfleisch, Franziska Greulich, Nina Henriette Uhlenhaut, and et al. 2021. "Cardioprotective Effects of Palmitoleic Acid (C16:1n7) in a Mouse Model of Catecholamine-Induced Cardiac Damage Are Mediated by PPAR Activation" International Journal of Molecular Sciences 22, no. 23: 12695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312695