
Skeletal Ryanodine Receptors Are Involved in Impaired Myogenic Differentiation in Duchenne Muscular Dystrophy Patients

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
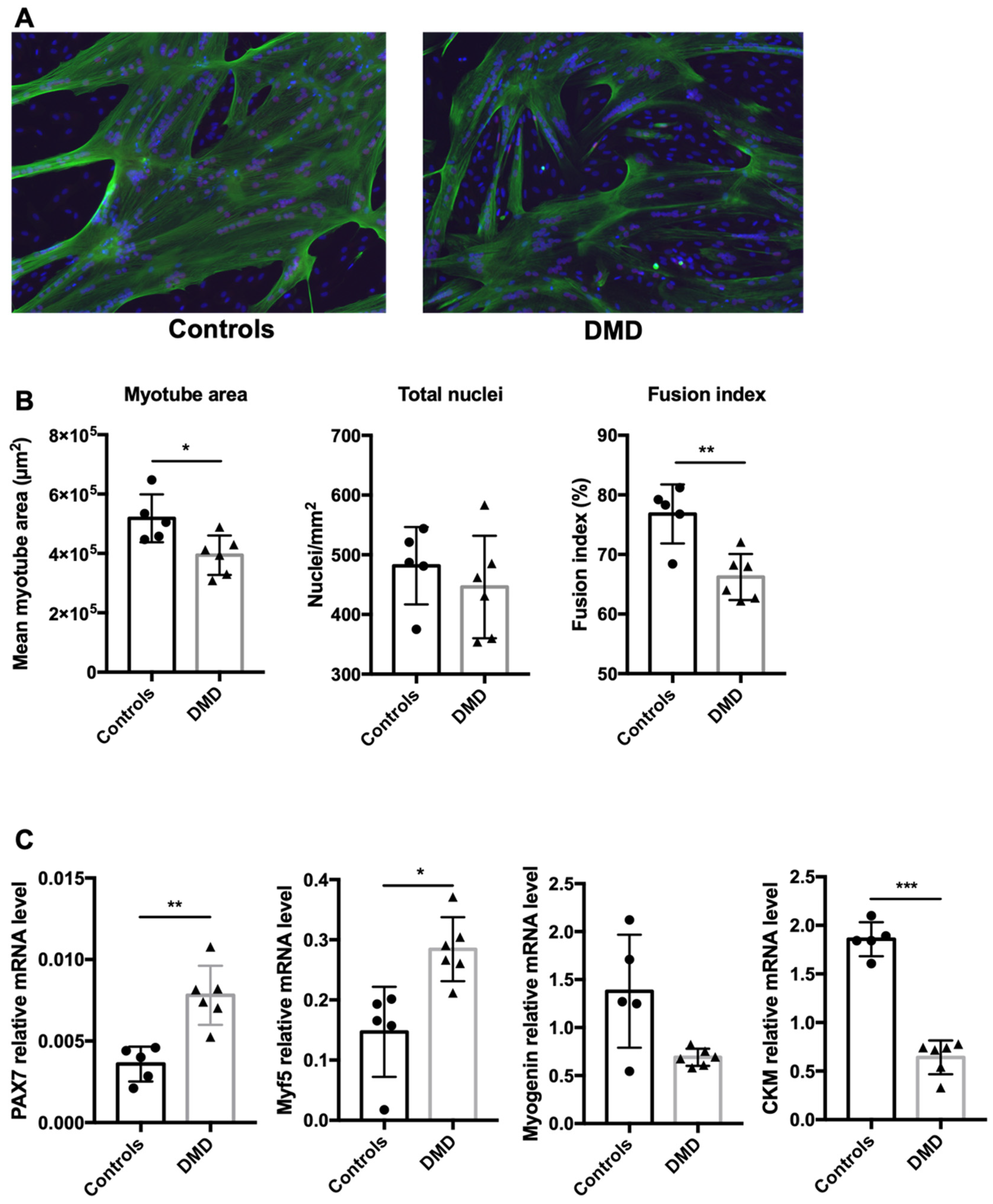
2.1. Myoblast Differentiation Is Delayed in DMD Patients
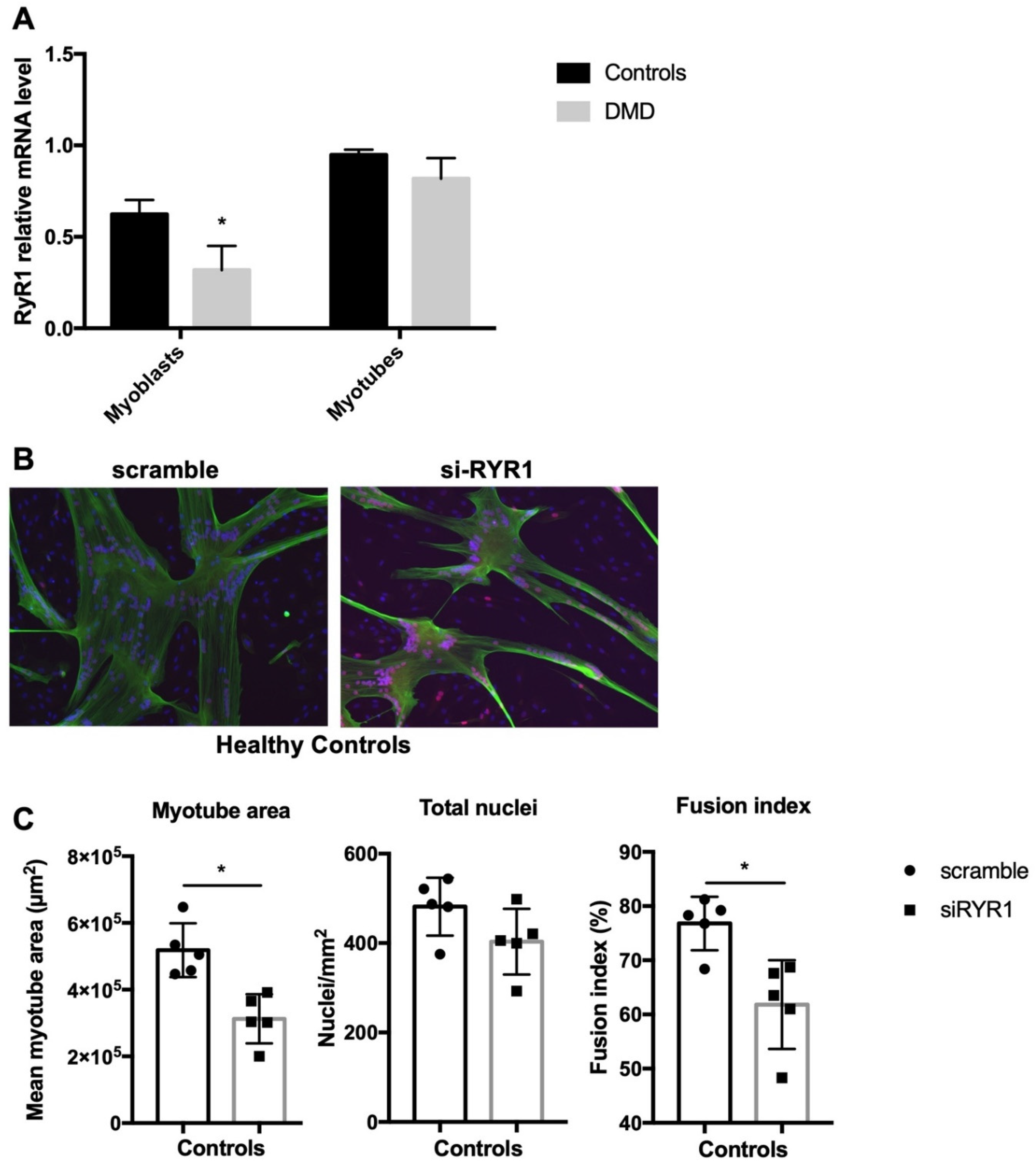
2.2. RYR1 Plays a Key Role during Early Human Myoblast Differentiation
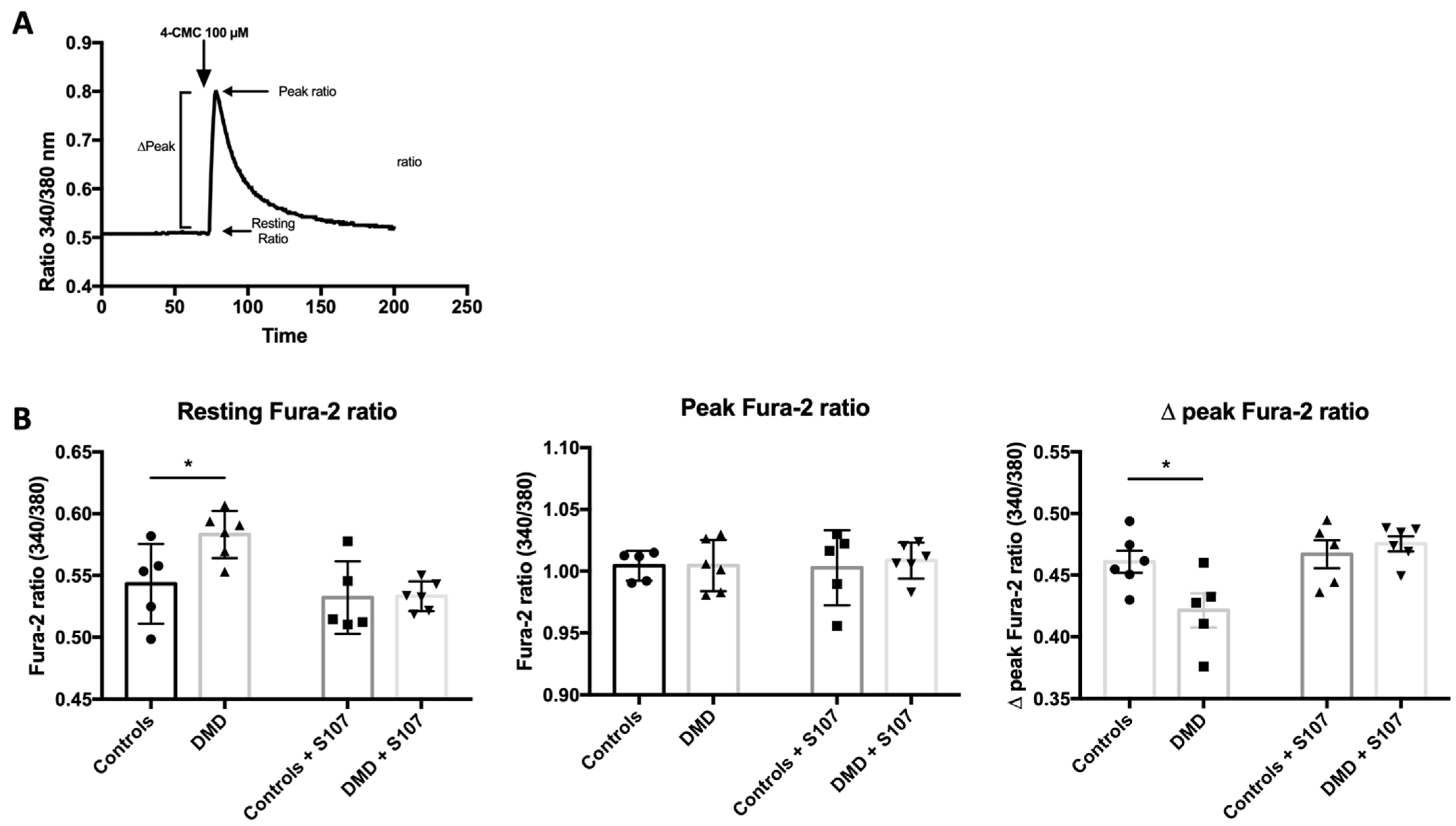
2.3. RYR1-Mediated Calcium Homeostasis Is Altered in DMD Myotubes
2.4. Calcium Leakage Is Improved by RYR1 Stabilizer S107 in DMD Myotubes
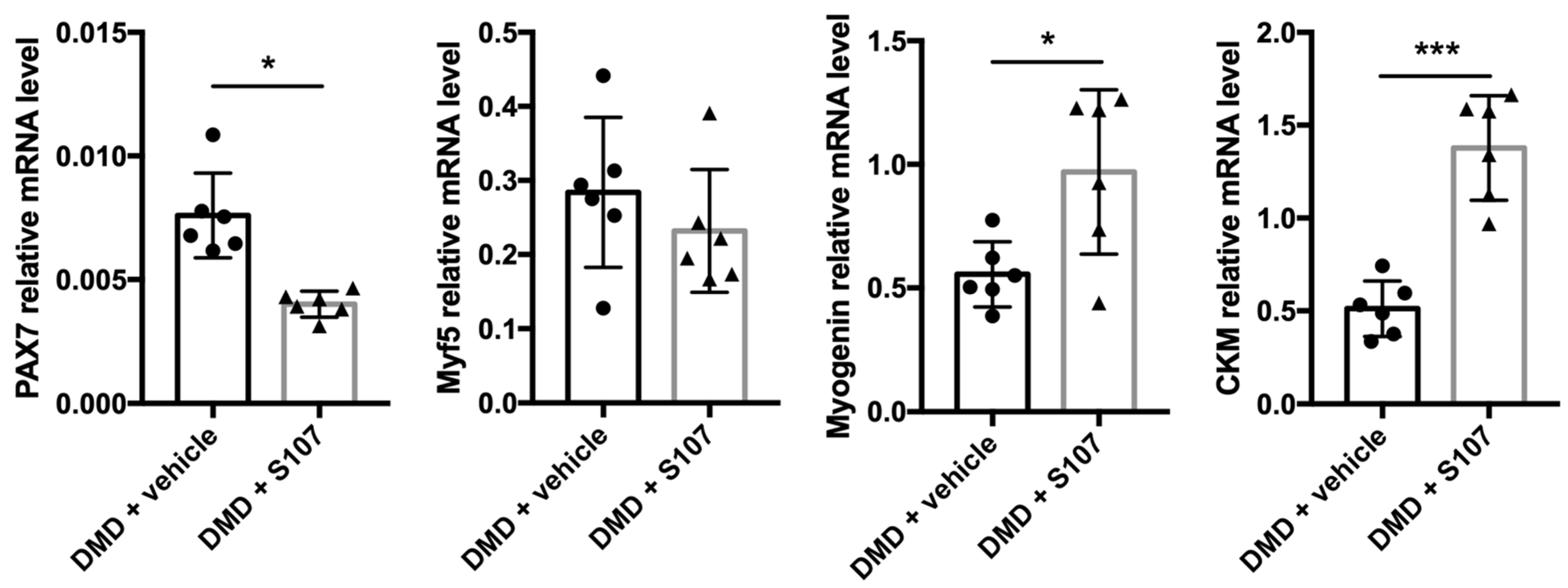
2.5. Inhibiting RYR1 Ca2+ Leak Improves Myogenic Differentiation in DMD Myoblasts
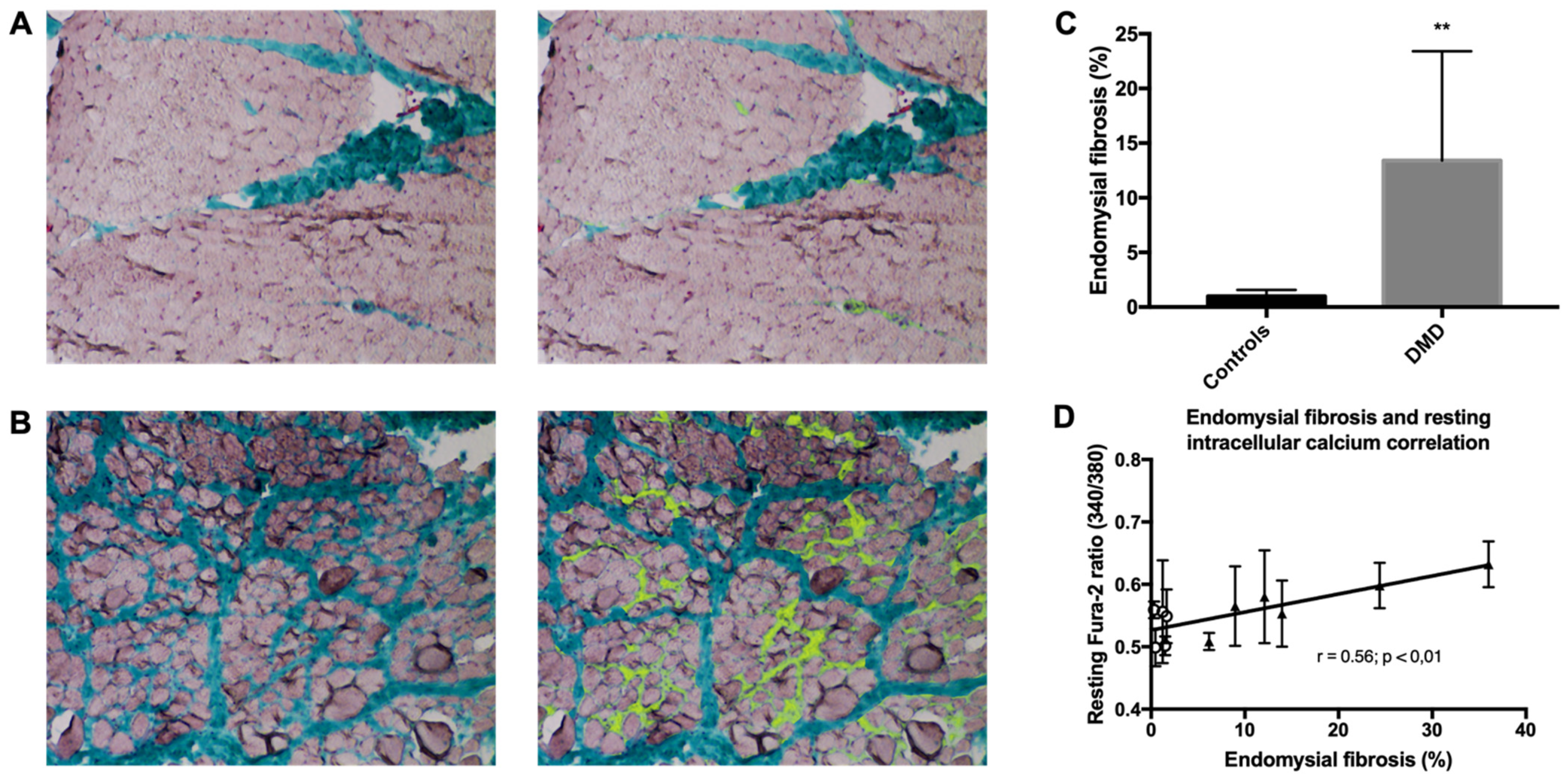
2.6. Endomysial Fibrosis Is Correlated with Elevated Intracellular Ca2+ Concentration
3. Discussion
4. Materials and Methods
4.1. Patients’ Samples
4.2. Histologic Diagnosis and Morphometric Analyses of Muscle Samples
4.3. Primary Human Skeletal Muscle Cell Culture
4.4. Transient si-RNA Transfections
4.5. Immunofluorescence Staining
4.6. Myotubes Area and Fusion Index Analyses
4.7. Reverse Transcription Quantitative PCR
4.8. Immunoprecipitation and Immunoblot Analyses
4.9. Measurement of Intracellular Ca2+ Variations
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deenen, J.C.W.; Horlings, C.G.C.; Verschuuren, J.J.G.M.; Verbeek, A.L.M.; van Engelen, B.G.M. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13–19. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [Green Version]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [Green Version]
- Alderton, J.M.; Steinhardt, R.A. How calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Trends Cardiovasc. Med. 2000, 10, 268–272. [Google Scholar] [CrossRef]
- Gailly, P. New aspects of calcium signaling in skeletal muscle cells: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta 2002, 1600, 38–44. [Google Scholar] [CrossRef]
- Ruegg, U.T.; Nicolas-Métral, V.; Challet, C.; Bernard-Hélary, K.; Dorchies, O.M.; Wagner, S.; Buetler, T.M. Pharmacological control of cellular calcium handling in dystrophic skeletal muscle. Neuromuscul. Disord. 2002, 12, S155–S161. [Google Scholar] [CrossRef]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.G.; Gervásio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010, 88, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bodensteiner, J.B.; Engel, A.G. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: A study of 567,000 muscle fibers in 114 biopsies. Neurology 1978, 28, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.Y.; Turner, P.R.; Denetclaw, W.F.; Steinhardt, R.A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 1990, 250, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Bertorini, T.E.; Cornelio, F.; Bhattacharya, S.K.; Palmieri, G.M.; Dones, I.; Dworzak, F.; Brambati, B. Calcium and magnesium content in fetuses at risk and prenecrotic Duchenne muscular dystrophy. Neurology 1984, 34, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.J.; Cao, J.H.; Deng, X.Q.; Zhang, C.L.; Qian, T.; Song, X.X.; Huang, B.S. Gene expression profiling of Duchenne muscular dystrophy reveals characteristics along disease progression. Genet. Mol. Res. 2014, 13, 1402–1411. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef] [Green Version]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.K.; Levin, J.B.; Hamilton, A.M.; Borodinsky, L.N. Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 2016, 59, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konig, S.; Béguet, A.; Bader, C.R.; Bernheim, L. The calcineurin pathway links hyperpolarization (Kir2.1)-induced Ca2+ signals to human myoblast differentiation and fusion. Development 2006, 133, 3107–3114. [Google Scholar] [CrossRef] [Green Version]
- Chin, E.R. The role of calcium and calcium/calmodulin-dependent kinases in skeletal muscle plasticity and mitochondrial biogenesis. Proc. Nutr. Soc. 2004, 63, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Stiber, J.A.; Tabatabaei, N.; Hawkins, A.F.; Hawke, T.; Worley, P.F.; Williams, R.S.; Rosenberg, P. Homer modulates NFAT-dependent signaling during muscle differentiation. Dev. Biol. 2005, 287, 213–224. [Google Scholar] [CrossRef]
- Constantin, B.; Cognard, C.; Raymond, G. Myoblast fusion requires cytosolic calcium elevation but not activation of voltage-dependent calcium channels. Cell Calcium 1996, 19, 365–374. [Google Scholar] [CrossRef]
- Dulhunty, A.F.; Wei-LaPierre, L.; Casarotto, M.G.; Beard, N.A. Core skeletal muscle ryanodine receptor calcium release complex. Clin. Exp. Pharmacol. Physiol. 2017, 44, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvaritch, E.; Depreux, F.; Kraeva, N.; Loy, R.E.; Goonasekera, S.A.; Boncompagni, S.; Boncompagi, S.; Kraev, A.; Gramolini, A.O.; Dirksen, R.T.; et al. An Ryr1I4895T mutation abolishes Ca2+ release channel function and delays development in homozygous offspring of a mutant mouse line. Proc. Natl. Acad. Sci. USA 2007, 104, 18537–18542. [Google Scholar] [CrossRef] [Green Version]
- Campbell, N.R.; Podugu, S.P.; Ferrari, M.B. Spatiotemporal characterization of short versus long duration calcium transients in embryonic muscle and their role in myofibrillogenesis. Dev. Biol. 2006, 292, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Oexle, K.; Kohlschütter, A. Cause of progression in Duchenne muscular dystrophy: Impaired differentiation more probable than replicative aging. Neuropediatrics 2001, 32, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Delaporte, C.; Dehaupas, M.; Fardeau, M. Comparison between the growth pattern of cell cultures from normal and Duchenne dystrophy muscle. J. Neurol. Sci 1984, 64, 149–160. [Google Scholar] [CrossRef]
- Jasmin, G.; Tautu, C.; Vanasse, M.; Brochu, P.; Simoneau, R. Impaired muscle differentiation in explant cultures of Duchenne muscular dystrophy. Lab. Investig. 1984, 50, 197–207. [Google Scholar]
- Melone, M.A.; Peluso, G.; Petillo, O.; Galderisi, U.; Cotrufo, R. Defective growth in vitro of Duchenne Muscular Dystrophy myoblasts: The molecular and biochemical basis. J. Cell. Biochem. 1999, 76, 118–132. [Google Scholar] [CrossRef]
- Merrick, D.; Stadler, L.K.J.; Larner, D.; Smith, J. Muscular dystrophy begins early in embryonic development deriving from stem cell loss and disrupted skeletal muscle formation. Dis. Model Mech. 2009, 2, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Andersson, D.C.; Meli, A.C.; Reiken, S.; Betzenhauser, M.J.; Umanskaya, A.; Shiomi, T.; D’Armiento, J.; Marks, A.R. Leaky ryanodine receptors in β-sarcoglycan deficient mice: A potential common defect in muscular dystrophy. Skelet. Muscle 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier, J.; Meli, A.C.; Thireau, J.; Roberge, S.; Shan, J.; Sassi, Y.; Reiken, S.R.; Rauzier, J.-M.; Marchand, A.; Chauvier, D.; et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc. Natl. Acad. Sci. USA 2011, 108, 13258–13263. [Google Scholar] [CrossRef] [Green Version]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [Green Version]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.-P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorzato, F.; Scutari, E.; Tegazzin, V.; Clementi, E.; Treves, S. Chlorocresol: An activator of ryanodine receptor-mediated Ca2+ release. Mol. Pharmacol. 1993, 44, 1192–1201. [Google Scholar] [PubMed]
- Herrmann-Frank, A.; Richter, M.; Sarközi, S.; Mohr, U.; Lehmann-Horn, F. 4-Chloro-m-cresol, a potent and specific activator of the skeletal muscle ryanodine receptor. Biochim. Biophys. Acta 1996, 1289, 31–40. [Google Scholar] [CrossRef]
- Herrmann-Frank, A.; Richter, M.; Lehmann-Horn, F. 4-Chloro-m-cresol: A specific tool to distinguish between malignant hyperthermia-susceptible and normal muscle. Biochem. Pharmacol. 1996, 52, 149–155. [Google Scholar] [CrossRef]
- Gafni, J.; Munsch, J.A.; Lam, T.H.; Catlin, M.C.; Costa, L.G.; Molinski, T.F.; Pessah, I.N. Xestospongins: Potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 1997, 19, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef]
- Schmalbruch, H. Regenerated muscle fibers in Duchenne muscular dystrophy: A serial section study. Neurology 1984, 34, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Sitzia, C.; Cassinelli, L.; Colleoni, F.; Parolini, D.; Giovanella, U.; Maciotta, S.; Colombo, A.; Meregalli, M.; Torrente, Y. Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling mediates delayed myogenesis in Duchenne muscular dystrophy fetal muscle. Development 2016, 143, 658–669. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, M.B.; Ribbeck, K.; Hagler, D.J.; Spitzer, N.C. A calcium signaling cascade essential for myosin thick filament assembly in Xenopus myocytes. J. Cell Biol. 1998, 141, 1349–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisaniello, A.; Serra, C.; Rossi, D.; Vivarelli, E.; Sorrentino, V.; Molinaro, M.; Bouché, M. The block of ryanodine receptors selectively inhibits fetal myoblast differentiation. J. Cell Sci. 2003, 116, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Lawal, T.A.; Todd, J.J.; Witherspoon, J.W.; Bönnemann, C.G.; Dowling, J.J.; Hamilton, S.L.; Meilleur, K.G.; Dirksen, R.T. Ryanodine receptor 1-related disorders: An historical perspective and proposal for a unified nomenclature. Skelet. Muscle 2020, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Arnaudeau, S.; Holzer, N.; Konig, S.; Bader, C.R.; Bernheim, L. Calcium sources used by post-natal human myoblasts during initial differentiation. J. Cell Physiol. 2006, 208, 435–445. [Google Scholar] [CrossRef]
- Gentil, C.; Leturcq, F.; Ben Yaou, R.; Kaplan, J.-C.; Laforet, P.; Pénisson-Besnier, I.; Espil-Taris, C.; Voit, T.; Garcia, L.; Piétri-Rouxel, F. Variable phenotype of del45-55 Becker patients correlated with nNOSμ mislocalization and RYR1 hypernitrosylation. Hum. Mol. Genet. 2012, 21, 3449–3460. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef] [PubMed]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef] [PubMed]
- Tjondrokoesoemo, A.; Li, N.; Lin, P.-H.; Pan, Z.; Ferrante, C.J.; Shirokova, N.; Brotto, M.; Weisleder, N.; Ma, J. Type 1 inositol (1,4,5)-trisphosphate receptor activates ryanodine receptor 1 to mediate calcium spark signaling in adult mammalian skeletal muscle. J. Biol. Chem. 2013, 288, 2103–2109. [Google Scholar] [CrossRef] [Green Version]
- Capogrosso, R.F.; Mantuano, P.; Uaesoontrachoon, K.; Cozzoli, A.; Giustino, A.; Dow, T.; Srinivassane, S.; Filipovic, M.; Bell, C.; Vandermeulen, J.; et al. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: Proof-of-concept study and independent validation of efficacy. FASEB J. 2017, 32, 1025–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, G.C.; Mokhonova, E.I.; Moran, M.; Sejbuk, N.E.; Wang, D.W.; Silva, O.; Wang, R.T.; Martinez, L.; Lu, Q.L.; Damoiseaux, R.; et al. Dantrolene Enhances Antisense-Mediated Exon Skipping in Human and Mouse Models of Duchenne Muscular Dystrophy. Sci. Transl. Med. 2012, 4, 164ra160. [Google Scholar] [CrossRef]
- Barthélémy, F.; Wang, R.T.; Hsu, C.; Douine, E.D.; Marcantonio, E.E.; Nelson, S.F.; Miceli, M.C. Targeting RYR Activity Boosts Antisense Exon 44 and 45 Skipping in Human DMD Skeletal or Cardiac Muscle Culture Models. Mol. Ther. Nucleic. Acids 2019, 18, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RYR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazorla, O.; Su, J.B.; Barthélémy, I.; Meli, A.C.; Chetboul, V.; Scheuermann, V.; Gouni, V.; Anglerot, C.; Richard, S.; Blot, S.; et al. Stabilizing ryanodine receptors improves left ventricular function in juvenile dogs with Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2021. accepted. [Google Scholar]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar]
- El Haddad, M.; Notarnicola, C.; Evano, B.; El Khatib, N.; Blaquière, M.; Bonnieu, A.; Tajbakhsh, S.; Hugon, G.; Vernus, B.; Mercier, J.; et al. Retinoic acid maintains human skeletal muscle progenitor cells in an immature state. Cell. Mol. Life Sci. 2017, 74, 1923–1936. [Google Scholar] [CrossRef]
- Stern-Straeter, J.; Bonaterra, G.A.; Hörmann, K.; Kinscherf, R.; Goessler, U.R. Identification of valid reference genes during the differentiation of human myoblasts. BMC Mol. Biol. 2009, 10, 66. [Google Scholar] [CrossRef] [Green Version]
- Gysembergh, A.; Lemaire, S.; Piot, C.; Sportouch, C.; Richard, S.; Kloner, R.A.; Przyklenk, K. Pharmacological manipulation of Ins(1,4,5)P3 signaling mimics preconditioning in rabbit heart. Am. J. Physiol. 1999, 277, H2458–H2469. [Google Scholar] [CrossRef]
- Virsolvy, A.; Farah, C.; Pertuit, N.; Kong, L.; Lacampagne, A.; Reboul, C.; Aimond, F.; Richard, S. Antagonism of Nav channels and α1-adrenergic receptors contributes to vascular smooth muscle effects of ranolazine. Sci. Rep. 2015, 5, 17969. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, P.; Notarnicola, C.; Meli, A.C.; Matecki, S.; Hugon, G.; Salvador, J.; Khalil, M.; Féasson, L.; Cances, C.; Cottalorda, J.; et al. Skeletal Ryanodine Receptors Are Involved in Impaired Myogenic Differentiation in Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2021, 22, 12985. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312985
Meyer P, Notarnicola C, Meli AC, Matecki S, Hugon G, Salvador J, Khalil M, Féasson L, Cances C, Cottalorda J, et al. Skeletal Ryanodine Receptors Are Involved in Impaired Myogenic Differentiation in Duchenne Muscular Dystrophy Patients. International Journal of Molecular Sciences. 2021; 22(23):12985. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312985
Chicago/Turabian StyleMeyer, Pierre, Cécile Notarnicola, Albano C. Meli, Stefan Matecki, Gérald Hugon, Jérémy Salvador, Mirna Khalil, Léonard Féasson, Claude Cances, Jérôme Cottalorda, and et al. 2021. "Skeletal Ryanodine Receptors Are Involved in Impaired Myogenic Differentiation in Duchenne Muscular Dystrophy Patients" International Journal of Molecular Sciences 22, no. 23: 12985. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312985