
Hippo Pathway in Regulating Drug Resistance of Glioblastoma

 and
and
Abstract
:
1. Glioblastoma Chemoresistance
2. Immunosuppressive Mechanisms in Glioblastoma
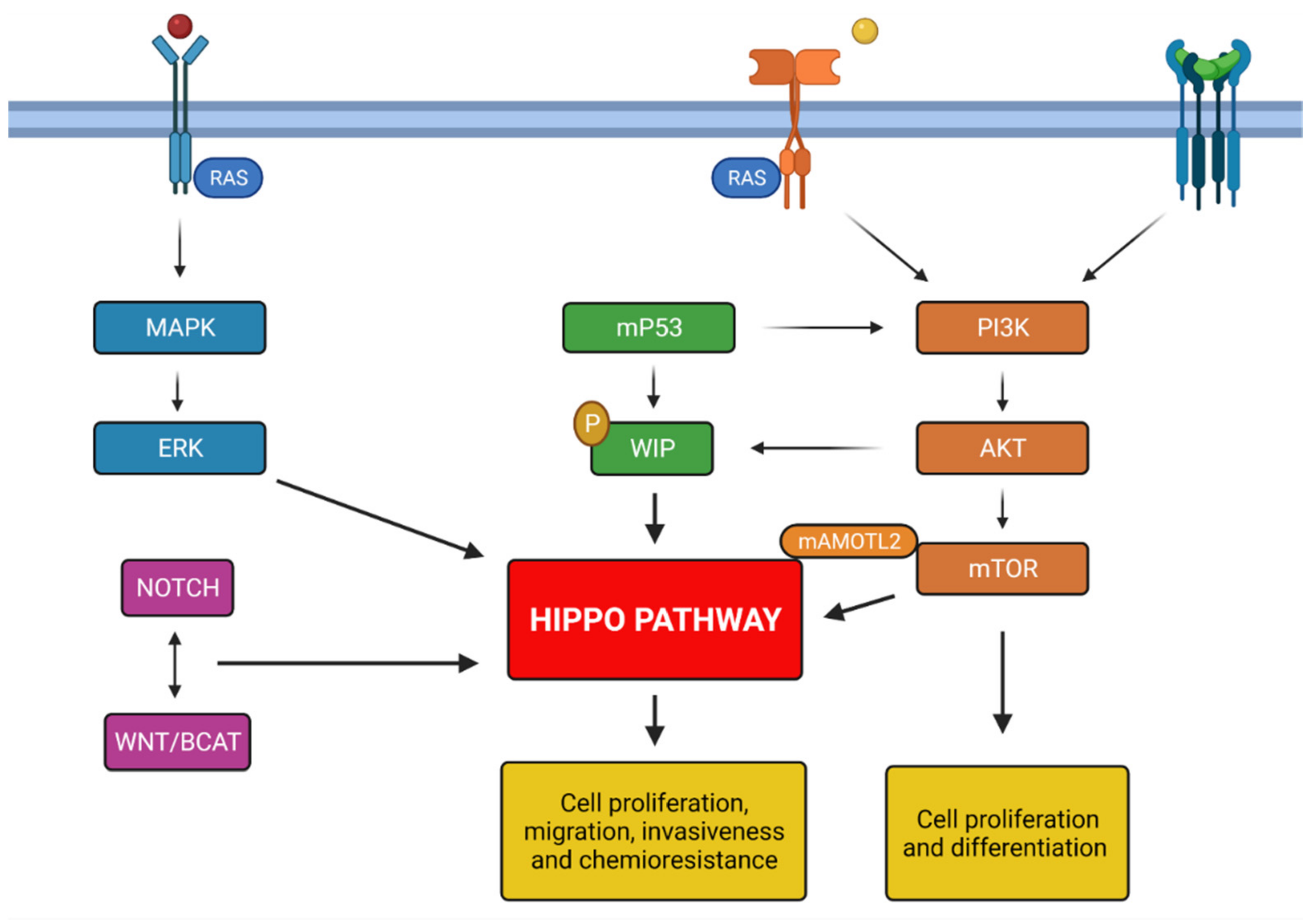
3. Pathways Involved in Glioblastoma Genesis
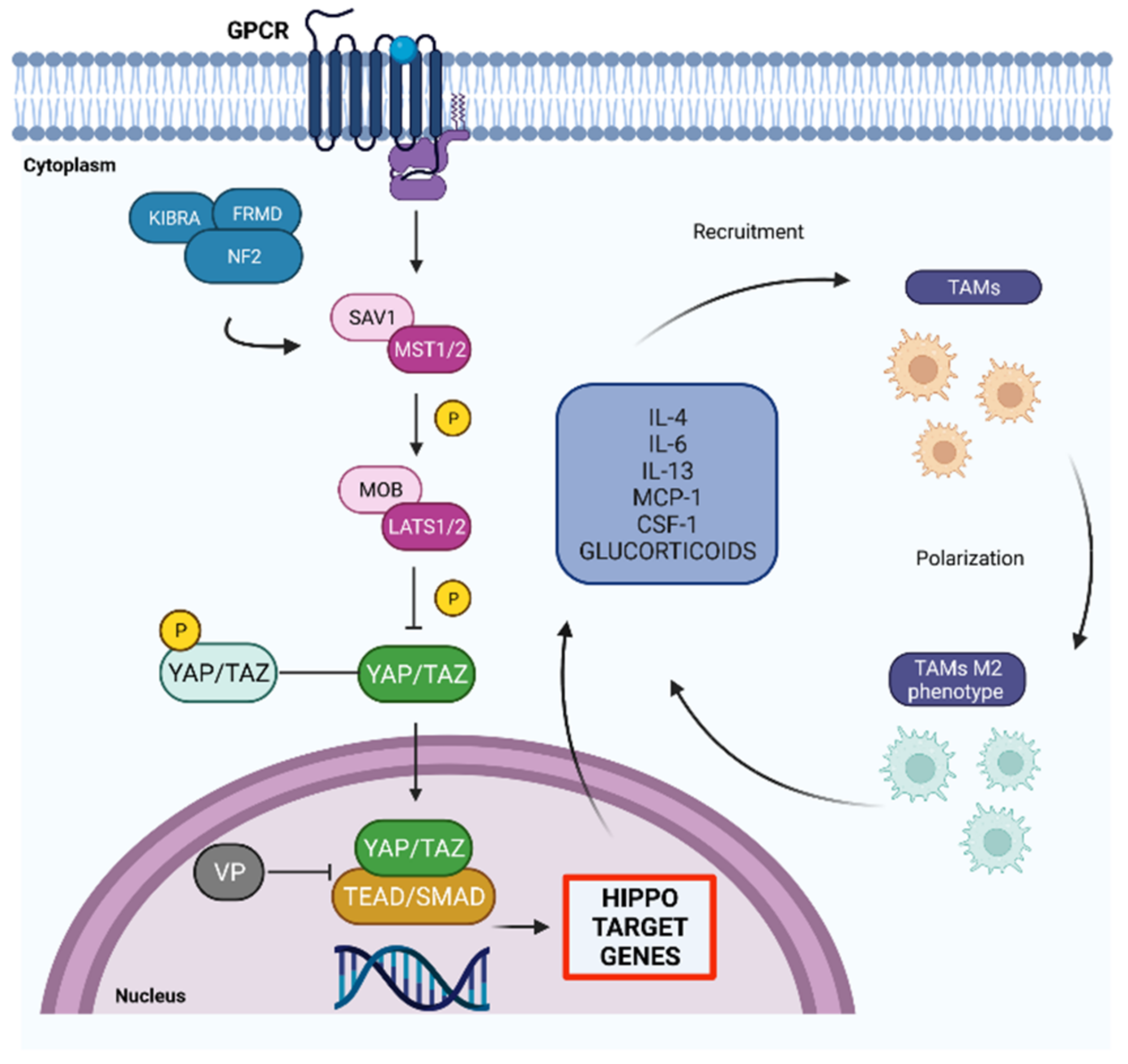
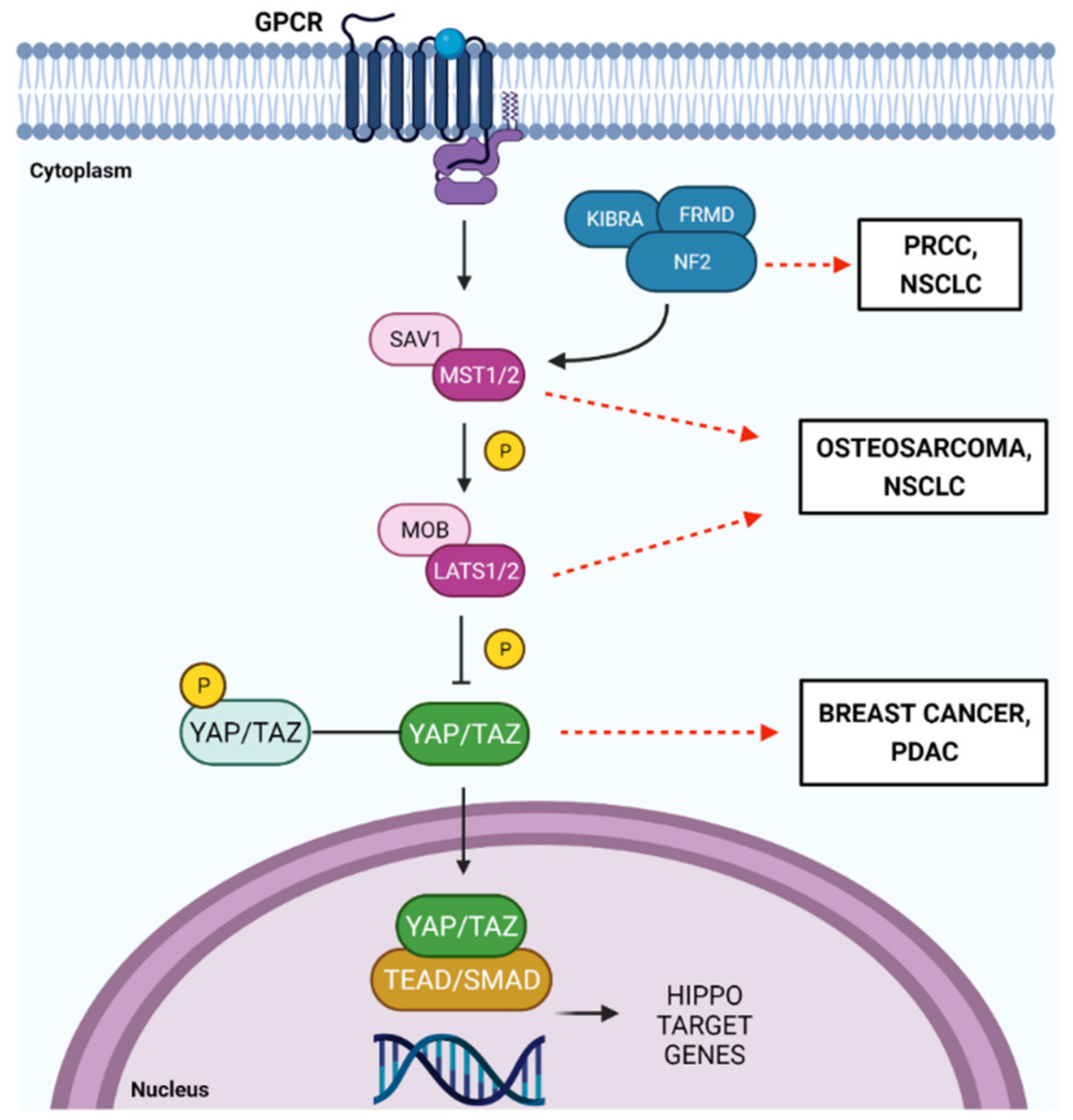
4. Background Hippo Pathway
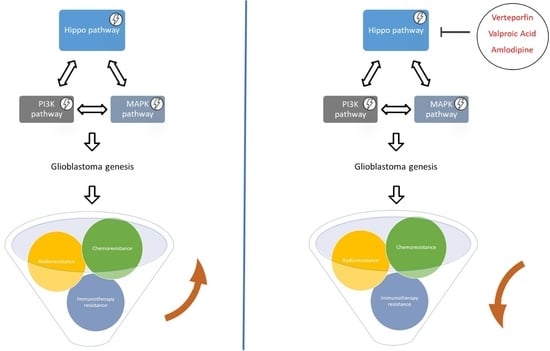
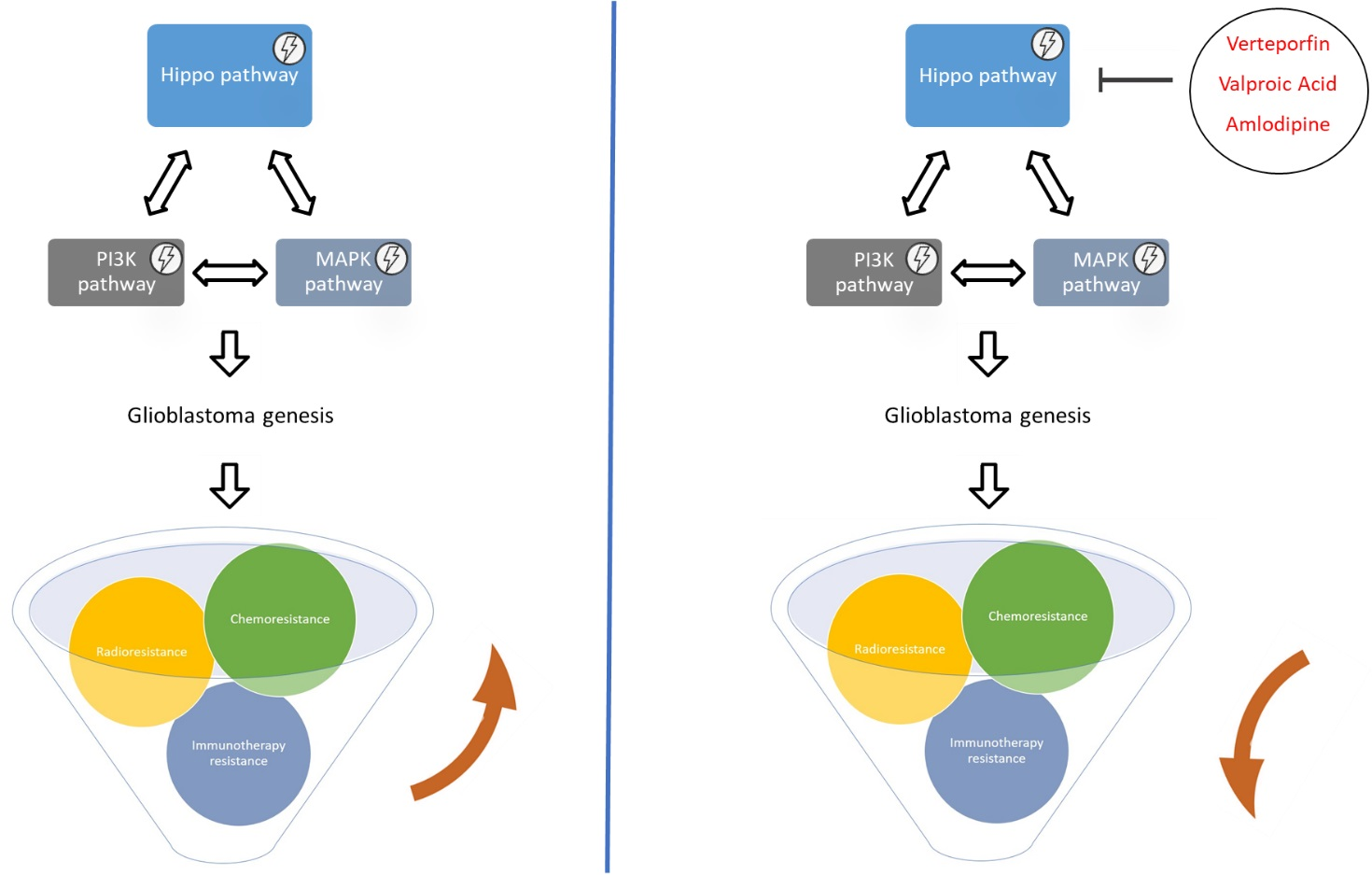
5. Hippo Pathway and Glioblastoma: Pharmacological Interventions
Funding
Conflicts of Interest
Abbreviations
| ALX | Amlexanox |
| AMOTL2 | angiomotin like-2 |
| BBB | blood brain barrier |
| BC | Breast Cancer |
| BZA | Bazedoxifene |
| CAR T | chimeric antigen receptor T cells modified |
| CNS | Central Nervous System |
| CSF-1 | colony stimulating factor-1 |
| CTGF | connective tissue growth factor |
| CTLs | cytotoxic T lymphocytes |
| Cyr61 | cysteine Rich angiogenic inducer 61 |
| D. | Drosophila |
| DDR | DNA damage response |
| Dox | Doxorubicin |
| DSBs | DNA double strand breaks |
| EGFR | epithelial growth factor receptor |
| EMT | epithelium-mesenchymal transition |
| FGF-1 | fibroblast growth factor |
| GBM | Glioblastoma |
| Hpo | Hippo |
| IDO | 3 dioxygenase |
| IKBKE | Inhibitor of nuclear factor Kappa-B Kinase |
| IR | Ionizing Radiation |
| LATS 1/2 | Large Tumor Suppressor homolog 1/2 |
| Mats | Mob As-Tumor-Suppressor |
| Mcp-1 | monocyte chemotactic protein 1 |
| miRNA | microRNA |
| MMPs | matrix metalloproteinases |
| MMR | mismatch repair |
| MOB1 A/B | Mps One Binder 1 |
| MST 1/2 | Mammalian Ste20-like 1 and 2 |
| NC | Nitidine chloride |
| PRCC | Papillary Renal Cell Carcinomas |
| NF2 | Neurofibromatosis type 2 |
| NRF2 | Nuclear Factor erythroid-derived 2-like 2 |
| NSCLC | Non-Small Cell Lung Cancer |
| PD-1 | programmed cell death protein-1 |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PD-L1 | programmed death-ligand 1 |
| PIP3 | PI-3-phosphate |
| RB | Retinoblastoma |
| Sav | Salvador |
| SERM | selective estrogen receptor modulator |
| TAMs | tumor associated macrophages |
| TAZ | Transcriptional Co-activator with PDZ-binding motif |
| TB | TEAD-Binding |
| TGF-b | Transforming Growth Factor beta |
| TMZ | Temozolomide |
| TPC | tumor promoting stem-like Characteristics |
| VPA | Valproic Acid |
| VP | Verteporfin |
| Wts | Warts |
| YAP | Yes-Associated Protein |
| Yki | Yorkie |
References
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Tian, T.; Li, A.; Lu, H.; Luo, R.; Zhang, M.; Li, Z. TAZ promotes temozolomide resistance by upregulating MCL-1 in human glioma cells. Biochem. Biophys. Res. Commun. 2015, 463, 638–643. [Google Scholar] [CrossRef]
- Zeng, H.; Yang, Z.; Xu, N.; Liu, B.; Fu, Z.; Lian, C.; Guo, H. Connective tissue growth factor promotes temozolomide resistance in glioblastoma through TGF-β1-dependent activation of Smad/ERK signaling. Cell Death Dis. 2017, 8, e2885. [Google Scholar] [CrossRef]
- Filppu, P.; Ramanathan, J.T.; Granberg, K.J.; Gucciardo, E.; Haapasalo, H.; Lehti, K.; Nykter, M.; Le Joncour, V.; Laakkonen, P. CD109-GP130 interaction drives glioblastoma stem cell plasticity and chemoresistance through STAT3 activity. JCI Insight 2021, 6, e141486. [Google Scholar] [CrossRef]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Zepeda, D.; Taghi, M.; Scherrmann, J.M.; Decleves, X.; Menet, M.C. ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas. Pharmaceutics 2020, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Dréan, A.; Rosenberg, S.; Lejeune, F.X.; Goli, L.; Nadaradjane, A.A.; Guehennec, J.; Schmitt, C.; Verreault, M.; Bielle, F.; Mokhtari, K.; et al. ATP binding cassette (ABC) transporters, Expression and clinical value in glioblastoma. J. Neuro-Oncol. 2018, 138, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declèves, X.; Amiel, A.; Delattre, J.Y.; Scherrmann, J.M.; Decleves, X.; Amiel, A.; Delattre, J.Y.; Scherrmann, J.M. Role of ABC Transporters in the Chemoresistance of Human Gliomas. Curr. Cancer Drug Targets 2006, 6, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20. [Google Scholar] [CrossRef]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Sheng, H.; Zhang, N.; Li, S.; Wei, X.; Zheng, X. Association of MSH6 mutation with glioma susceptibility, drug resistance and progression. Mol. Clin. Oncol. 2016, 5, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yee, P.P.; Wei, Y.; Liu, Z.; Kawasawa, Y.I.; Li, W. Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J. Cell Sci. 2019, 132, jcs225714. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaréa, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Y.; Zhou, D.; Wang, K.; Wang, X.; Wang, X.; Jiang, Y.; Zhao, M.; Yu, R.; Zhou, X. Radiation-induced YAP activation confers glioma radioresistance via promoting FGF2 transcription and DNA damage repair. Oncogene 2021, 40, 4580–4591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, P.; Wang, Z.; Zhang, Y.; Xie, P.; Geng, D.; Jiang, Y.; Yu, R.; Zhou, X. β-catenin-mediated YAP signaling promotes human glioma growth. J. Exp. Clin. Cancer Res. 2017, 36, 1–11. [Google Scholar] [CrossRef]
- Kim, S.; Jho, E.H. Merlin, a regulator of Hippo signaling, regulates Wnt/β-catenin signaling. BMB Rep. 2016, 49, 357–358. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zhou, L.; Han, N.; Zhang, M.; Lyu, X. Wnt/β-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther Onkol. 2015, 191, 672–680. [Google Scholar] [CrossRef]
- Cammarata, F.P.; Torrisi, F.; Forte, G.I.; Minafra, L.; Bravatà, V.; Pisciotta, P.; Savoca, G.; Calvaruso, M.; Petringa, G.; Cirrone, G.A.P.; et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int. J. Mol. Sci. 2019, 20, 4745. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.H.; Bi, J.F.; Cloughesy, T.; Cavenee, W.K.; Mischel, P.S. Emerging function of mTORC2 as a core regulator in glioblastoma, Metabolic reprogramming and drug resistance. Cancer Biol. Med. 2014, 11, 255–263. [Google Scholar]
- Artinian, N.; Cloninger, C.; Holmes, B.; Benavides-Serrato, A.; Bashir, T.; Gera, J. Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness. J. Biol. Chem. 2015, 290, 19387–19401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Kumar, S.; Nishimura, R.N.; Gera, J. mTORC2-mediated direct phosphorylation regulates YAP activity promoting glioblastoma growth and invasive characteristics. Neoplasia 2021, 23, 951–965. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. Mgmt gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Schackert, G.; Yaya-Tur, R.; Rojas-Marcos, I.; Herman, J.G.; Esteller, M. Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2007, 83, 91–93. [Google Scholar] [CrossRef]
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841. [Google Scholar]
- Giunti, L.; Da Ros, M.; Vinci, S.; Gelmini, S.; Iorio, A.L.; Buccoliero, A.M.; Cardellicchio, S.; Castiglione, F.; Genitori, L.; De Martino, M.; et al. Anti-miR21 oligonucleotide enhances chemosensitivity of t98g cell line to doxorubicin by inducing apoptosis. Am. J. Cancer Res. 2015, 5, 231–242. [Google Scholar]
- Bai, Y.; Liao, H.; Liu, T.; Zeng, X.; Xiao, F.; Luo, L.; Guo, H.; Guo, L. Mir-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (EAG1). Eur. J. Cancer. 2013, 49, 710–724. [Google Scholar] [CrossRef]
- Qian, Z.; Zhou, S.; Zhou, Z.; Yang, X.; Que, S.; Lan, J.; Qiu, Y.; Lin, Y. Mir-146b-5p suppresses glioblastoma cell resistance to temozolomide through targeting TRAF6. Oncol. Rep. 2017, 38, 2941–2950. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Mingyi, M.; Qiu, X.; Qiu, Y. MicroRNA-101 reverses temozolomide resistance by inhibition of GSK3β in glioblastoma. Oncotarget 2016, 7, 79584–79595. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer. 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Visone, R.; Croce, C.M. miRNAs and cancer. Am. J. Pathol. 2009, 174, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A MicroRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer–A brief overview. Adv. Biol. Regul. 2015, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; Greco, S.J.; Rameshwar, P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015, 6, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Feng, R.; Dong, L. Knockdown of MicroRNA-127 reverses adriamycin resistance via cell cycle arrest and apoptosis sensitization in adriamycin-resistant human glioma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6107–6116. [Google Scholar]
- Blower, P.E.; Chung, J.H.; Verducci, J.S.; Lin, S.; Park, J.K.; Dai, Z.; Liu, C.G.; Schmittgen, T.D.; Reinhold, W.C.; Croce, C.M.; et al. MicroRNAs modulate the chemosensitivity of tumor cells. Mol. Cancer Ther. 2008, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/PGP-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Lu, X.; Cao, Y. MicroRNA and Signal Transduction Pathways in Tumor Radiation Response. Cell Signal 2013, 25, 1625–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taghi, M.; Toossi, B.; Dolat, E.; Khanbabaei, H.; Zafari, N.; Azimian, H. microRNAs, Potential glioblastoma radiosensitizer by targeting radiation-related molecular pathways. Mutat. Res. 2019, 816–818, 111679. [Google Scholar]
- Szatkowska, M.; Krupa, R. Regulation of DNA Damage Response and Homologous Recombination Repair by microRNA in Human Cells Exposed to Ionizing Radiation. Cancers 2020, 12, 1838. [Google Scholar] [CrossRef]
- Natarajan, V. Regulation of DNA repair by non-coding miRNAs. Noncoding RNA Res. 2016, 1, 64–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles, Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zocco, D.; Ferruzzi, P.; Cappello, F.; Kuo, W.P.; Fais, S. Extracellular vesicles as shuttles of tumor biomarkers and anti-tumor drugs. Front. Oncol. 2014, 4, 267. [Google Scholar] [CrossRef] [Green Version]
- Antonyak, M.A.; Cerione, R.A. Microvesicles as mediators of intercellular communication in cancer. Methods Mol. Biol. 2014, 1165, 147–173. [Google Scholar]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes, Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- Namee, N.M.C.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cell. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Zeng, A.L.; Yan, W.; Liu, Y.W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381. [Google Scholar] [CrossRef] [Green Version]
- De Los Santos, M.C.; Dragomir, M.P.; Calin, G.A. The role of exosomal long non-coding RNAs in cancer drug resistance. Cancer Drug Resist. 2019, 2, 1178–1192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yin, J.; Lu, C.; Wei, Y.; Zeng, A.; You, Y. Exosomal transfer of long non-coding RNA SBF2-AS1 enhances chemoresistance to temozolomide in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 166. [Google Scholar] [CrossRef]
- Shang, C.; Tang, W.; Pan, C.; Hu, X.; Hong, Y. Long non-coding RNA TUSC7 inhibits temozolomide resistance by targeting miR-10a in glioblastoma. Cancer Chemother. Pharmacol. 2018, 81, 671–678. [Google Scholar] [CrossRef]
- Lu, C.; Wei, Y.; Wang, X.; Zhang, Z.; Yin, J.; Li, W.; Chen, L.; Lyu, X.; Shi, Z.; Yan, W.; et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol. Cancer 2020, 19, 28. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Xu, N.; Liu, Y.; Liu, B.; Yang, Z.; Fu, Z.; Lian, C.; Guo, H. Genomic profiling of long non-coding RNA and mRNA expression associated with acquired temozolomide resistance in glioblastoma cells. Int. J. Oncol. 2017, 51, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomé, M.; Tchorz, J.; Gassmann, M.; Bettler, B. Constitutive activation of Notch2 signalling confers chemoresistance to neural stem cells via transactivation of fibroblast growth factor receptor-1. Stem Cell Res. 2019, 35, 101390. [Google Scholar] [CrossRef] [PubMed]
- Alafate, W.; Xu, D.; Wu, W.; Xiang, J.; Ma, X.; Xie, W.; Bai, X.; Wang, M.; Wang, J. Loss of PLK2 induces acquired resistance to temozolomide in GBM via activation of notch signaling. J. Exp. Clin. Cancer Res. 2020, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front Cell Dev. Biol. 2021, 9, 650772. [Google Scholar] [CrossRef]
- Hao, B.; Chen, X.; Cao, Y. Yes-associated protein 1 promotes the metastasis of U251 glioma cells by upregulating Jagged-1 expression and activating the Notch signal pathway. Exp. Ther. Med. 2018, 16, 1411–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma–Are we there yet? Neuro-Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Gramatzki, D.; Dehler, S.; Rushing, E.J.; Zaugg, K.; Hofer, S.; Yonekawa, Y.; Bertalanffy, H.; Valavanis, A.; Korol, D.; Rohrmann, S.; et al. Glioblastoma in the canton of Zurich, Switzerland revisited, 2005 to 2009. Cancer 2016, 122, 2206–2215. [Google Scholar] [CrossRef]
- Jackson, C.M.; Kochel, C.M.; Nirschl, C.J.; Durham, N.M.; Ruzevick, J.; Alme, A.; Francica, B.J.; Elias, J.; Daniels, A.; Dubensky, T.W.; et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin. Cancer Res. 2016, 22, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Jähnisch, H.; Füssel, S.; Kiessling, A.; Wehner, R.; Zastrow, S.; Bachmann, M.; Rieber, E.P.; Wirth, M.P.; Schmitz, M. Dendritic cell-based immunotherapy for prostate cancer. Clin. Dev. Immunol. 2010, 2010, 517493. [Google Scholar] [CrossRef]
- Aurelian, L. Oncolytic viruses as immunotherapy, Progress and remaining challenges. OncoTargets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef] [Green Version]
- Jena, B.; Dotti, G.; Cooper, L.J. Redirecting t-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 2010, 116, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13R_2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Yang, S.; Zhang, F.; Cheng, F.; Wang, X.; Rao, J. Influence of the Hippo-YAP signaling pathway on tumor associated macrophages (TAMs) and its implications on cancer immunosuppressive microenvironment. Ann. Transl. Med. 2020, 8, 399. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Xiong, B. M2 macrophages confer resistance to 5-fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling. OncoTargets Ther. 2019, 12, 3051–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumor-infiltrating macrophages via CCL2/CCR2 signaling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumor-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer, A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Clin. Trial Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Helmut, K. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Zhou, T.-Y.; Zhou, Y.-L.; Qian, M.-J.; Fang, Y.-Z.; Ye, S.; Xin, W.-X.; Yang, W.-C.; Wu, H.-H. Interleukin-6 induced by YAP in hepatocellular carcinoma cells recruits tumor-associated macrophages. J. Pharmacol. Sci. 2018, 138, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Yang, C.K.; Wei, P.L.; Huynh, T.T.; Whang-Peng, J.; Meng, T.C.; Hsiao, M.; Tzeng, Y.M.; Wu, A.T.; Yen, Y. Ovatodiolide suppresses colon tumorigenesis and prevents polarization of M2 tumor-associated macrophages through YAP oncogenic pathways. J. Hematol. Oncol. 2017, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-induced PD-L1 expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol. Res. 2018, 6, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance, The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor VIII as a molecular target in cancer therapy. Endocr. Relat. Cancer 2001, 8, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-mutant Glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569. [Google Scholar] [CrossRef]
- Feldkamp, M.M.; Lala, P.; Lau, N.; Roncari, L.; Guha, A. Expression of activated epidermal growth factor receptors, Rasguanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999, 45, 1442–1453. [Google Scholar] [CrossRef]
- Barrette, A.M.; Tome-Garcia, J.; Zaware, N.; Zhou, M.M.; Birtwistle, M.; Tsankova, N. Verteporfin treatment inhibits GBM growth and migration and informs Hippo/RTK crosstalk. Neuro-Oncol. 2018, 20, vi89. [Google Scholar] [CrossRef] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.Y.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.W.; Lim, C.J.; Chong, Y.F.; Pobbati, A.V.; Huang, C.X.; Hong, W.J. Hippo Pathway-independent Restriction of TAZ and YAP by Angiomotin. J. Biol. Chem. 2011, 286, 7018–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699703. [Google Scholar] [CrossRef]
- Nakada, M. Aberrant signaling pathways in glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [Green Version]
- Munro, S.; Carr, S.M.; La Thangue, N.B. Diversity within the pRb pathway: Is there a code of conduct? Oncogene 2012, 31, 4343–4352. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway, positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Raj, N.; Bam, R. Reciprocal Crosstalk between YAP1/Hippo Pathway and the p53 Family Proteins, Mechanisms and Outcomes in Cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. ATM regulates a RASSF1A-dependent DNA damage response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Weyden, L.; Papaspyropoulos, A.; Poulogiannis, G.; Rust, A.G.; Rashid, M.; Adams, D.J.; Arends, M.J.; O’Neill, E. Loss of RASSF1A synergizes with deregulated RUNX2 signaling in tumorigenesis. Cancer Res. 2012, 72, 3817–3827. [Google Scholar] [CrossRef] [Green Version]
- Pefani, P.E.; O’Neill, E. Hippo pathway and protection of genome stability in response to DNA damage. FEBS J. 2016, 283, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Kim, M.; Jho, E.H. Wnt/beta-catenin signaling: From plasma membrane to nucleus. Biochem. J. 2013, 450, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-beta signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Ouyang, T.; Meng, W.; Li, M.; Hong, T.; Zhang, N. Recent Advances of the Hippo/YAP Signaling Pathway in Brain Development and Glioma. Cell. Mol. Neurobiol. 2020, 4, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. Neuromolecular Med. 2018, 20, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.K.; Ahn, S.H.; Lee, J.; Nam, D.H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2016, 96, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-like Cells. Cancer Res. 2017, 77, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Suwala, A.K.; Hanaford, A.; Kahlert, U.D.; Maciaczyk, J. Clipping the wings of glioblastoma, Modulation of Wnt as a novel therapeutic strategy. J. Neuropathol. Exp. Neurol. 2016, 75, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jho, E.H. Cross talk between Wnt/β-catenin and Hippo signaling pathways: A brief review. BMB Rep. 2014, 47, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, K.; Reddy, S.P.; Vinnakota, K.; Britto, R.; Subbarayan, M.; Nambiar, S.; Hebbar, A.; Samuel, C.; Shetty, M.; Sreepathi, H.K.; et al. Upregulation of ASCL1 and inhibition of Notch signaling pathway characterize progressive astrocytoma. Oncogene 2005, 24, 7073–7083. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Gersey, Z.; Osiason, A.D.; Bloom, L.; Shah, S.; Thompson, J.W.; Bregy, A.; Agarwal, N.; Komotar, R.J. Therapeutic targeting of the notch pathway in glioblastoma multiforme. World Neurosurg. 2019, 131, 252–263.e2. [Google Scholar] [CrossRef] [PubMed]
- Conlon, I.; Raff, M. Size control in animal development. Cell 1999, 96, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.; Tapon, N. The Salvador-Warts-Hippo pathway-an emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Han, H.; Seo, G.; Vargas, R.E.; Yang, B.; Chuc, K.; Zhao, H.; Wang, W. Systematic analysis of the Hippo pathway organization and oncogenic alteration in evolution. Sci. Rep. 2020, 10, 3173. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Kango-Singh, M.; Nolo, R.; Tao, C.; Verstreken, P.; Hiesinger, P.R.; Bellen, H.J.; Halderet, G. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 2002, 129, 5719–5730. [Google Scholar] [CrossRef] [Green Version]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.R.; Schiripo, T.A.; Haber, D.A.; Hariharanet, I.K. Salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Pantalacci, S.; Tapon, N.; Leopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003, 5, 921–927. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, R.; Chen, L.F.; Shigesada, K.; Murakami, Y.; Ito, Y. A WW domain-containing yes-associated protein (YAP) is a novel tran-scriptional co-activator. EMBO J. 1999, 18, 2551–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.K.; Sullivan, A.J.; Medina, R.; Ito, Y.; Van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004, 23, 790–799. [Google Scholar] [CrossRef]
- Van Rensburg, H.J.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes. Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical inter-action with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.N.; Olson, E.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Heng, B.C.; Zhang, X.; Aubel, D.; Bai, Y.; Li, X.; Wei, Y.; Fussenegger, M.; Deng, X. Role of YAP/TAZ in Cell Lineage Fate Determination and Related Signaling Pathways Front Cell. Dev. Biol. 2020, 8, 735. [Google Scholar]
- Karaman, R.; Halder, R. Cell Junctions in Hippo Signaling. Cold Spring Harb. Perspect. Biol. 2018, 10, a028753. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signaling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Wada, K.I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; Vocke, C.D.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. Cancer Genome Atlas Research Network. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar]
- Sourbier, C.; Liao, P.J.; Ricketts, C.J.; Wei, D.; Yang, Y.; Baranes, S.M.; Gibbs, B.K.; Ohanjanian, L.; Krane, L.S.; Scroggins, B.T.; et al. Targeting loss of the Hippo signaling pathway in NF2-deficient papillary kidney cancers. Oncotarget 2018, 9, 10723–10733. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Zhang, H.; Chong, Y.; Guan, B.; Guo, P. YAP Promotes VEGFA Expression and Tumor Angiogenesis Though Gli2 in Human Renal Cell Carcinoma. Biomedical 2019, 50, 225–233. [Google Scholar] [CrossRef]
- Cao, J.J.; Zhao, X.M.; Wang, D.L.; Chen, K.H.; Sheng, X.; Li, W.B.; Li, M.C.; Liu, W.J.; He, J. YAP is overexpressed in clear cell renal cell carcinoma and its knockdown reduces cell proliferation and induces cell cycle arrest and apoptosis. Oncol. Rep. 2014, 32, 1594–1600. [Google Scholar] [CrossRef] [Green Version]
- Bielack, S.S.; Kempf-Bielack, B.; Delling, G.; Exner, G.U.; Flege, S.; Helmke, K.; Kotz, R.; Salzer-Kuntschik, M.; Werner, M.; Winkelmann, W.; et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: An analysis of 1702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J. Clin. Oncol. 2002, 20, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Wu, Y.N.; Huang, J.Q.; Wang, W.; Xu, M.; Jia, J.P.; Han, G.; Mao, B.B.; Bi, W.Z. Hippo/YAP signaling pathway is involved in osteosarcoma chemoresistance. Chin. J. Cancer. 2016, 35, 47. [Google Scholar] [CrossRef] [Green Version]
- Zaman, A.; Bivona, T.G. Emerging application of genomics-guided therapeutics in personalized lung cancer treatment. Ann. Transl. Med. 2018, 6, 160. [Google Scholar] [CrossRef]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs), Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef] [Green Version]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Gobbi, G.; Donati, B.; Do Valle, I.F.; Reggiani, F.; Torricelli, F.; Remondini, D.; Castellani, G.; Ambrosetti, D.C.; Ciarrocchi, A.; Sancisi, V. The Hippo pathway modulates resistance to BET proteins inhibitors in lung cancer cells. Oncogene 2019, 38, 6801–6817. [Google Scholar] [CrossRef]
- Lau, A.N.; Curtis, S.J.; Fillmore, C.M.; Rowbotham, S.P.; Mohseni, M.; Wagner, D.E.; Beede, A.M.; Montoro, D.T.; Sinkevicius, K.W.; Walton, Z.E.; et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014, 33, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Lo Sardo, F.; Strano, S.; Blandino, G. YAP and TAZ in lung cancer: Oncogenic role and clinical targeting. Cancers 2018, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Torres, C.; Grippo, P.J. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann. Med. 2018, 50, 277–287. [Google Scholar] [CrossRef]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Cui, J.; Xia, T.; Jia, Z.; Wang, L.; Wei, W.; Zhu, A.; Gao, Y.; Xie, K.; Quan, M. Hippo transducer TAZ promotes epithelial—Mesenchymal transition and supports pancreatic cancer progression. Oncotarget 2015, 6, 35949–35963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diep, C.H.; Zucker, K.M.; Hostetter, G.; Watanabe, A.; Hu, C.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Down-regulation of YES- associated protein 1 expression reduces cell proliferation and clonogenicity of pancreatic cancer cells. PLoS ONE 2012, 7, e32783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcedo Allende, M.T.; Zeron-Medina, J.; Hernandez, J.; Macarulla, T.; Balsells, J.; Merino, X.; Allende, H.; Tabernero, J.; Ramon, Y.C.S. Overexpression of YES-associated protein 1, an independent prognostic marker in patients with pancreatic ductal adenocarcinoma, correlated with liver metastasis and poor prognosis. Pancreas 2017, 46, 913–920. [Google Scholar] [CrossRef]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal 2014, 7, ra42. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer; A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Li, D.; Li, H.; Wang, L.; Tian, G.; Dong, Y. YAP overexpression promotes the epithelia–-mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol. Med. Rep. 2016, 13, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Wang, M.; Xu, S.; Guo, X.; Jiang, J. Upregulation of mir-181c contributes to chemoresistance in pancreatic cancer by inactivating the Hippo signaling pathway. Oncotarget 2015, 6, 44466–44479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.A.; Wang, R.; Miao, J.; Oliva, E.; Shen, X.; Wheeler, T.; Hilsenbeck, S.G.; Orsulic, S.; Goode, S. Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 2010, 70, 8517–8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Lu, J.; Zhang, F.; Li, H.; Zhang, B.; Wu, X.; Tan, Z.; Zhang, L.; Gao, G.; Mu, J.; et al. Yes-associated protein 1 (YAP1) promotes human gallbladder tumor growth via activation of the AXL/MAPK pathway. Cancer Lett. 2014, 355, 201–209. [Google Scholar] [CrossRef]
- Avruch, J.; Zhou, D.; Bardeesy, N. YAP oncogene overexpression supercharges colon cancer proliferation. Cell Cycle 2012, 11, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Di Benedetto, A.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, Y.; Luo, J.; Zheng, J.; Shao, G. YAP1 overexpression is associated with poor prognosis of breast cancer patients and induces breast cancer cell growth by inhibiting PTEN. FEBS Open Bio 2019, 9, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Lei, Q.Y. Regulation of TAZ in cancer. Protein Cell 2016, 7, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Wang, Z.; Huang, W.; Lei, Q.Y. G protein-coupled receptors: Bridging the gap from the extracellular signals to the Hippo pathway. Acta Biochim. Biophys. Sin. 2015, 47, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer. 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Namani, A.; Cui, Q.Q.; Wu, Y.; Wang, H.; Wang, X.J.; Tang, X. NRF2-regulated metabolic gene signature as a prognostic biomarker in non-small cell lung cancer. Oncotarget 2017, 8, 69847–69862. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Cong, Z.X.; Wang, H.D.; Wang, J.W.; Zhou, Y.; Pan, H.; Zhang, D.D.; Zhu, L. ERK and PI3K signaling cascades induce Nrf2 activation and regulate cell viability partly through Nrf2 in human glioblastoma cells. Oncol. Rep. 2013, 30, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, M.; Higa, T.; Sonoda, Y.; Murakami, S.; Dodo, M.; Kitamura, H.; Taguchi, K.; Shibata, T.; Watanabe, M.; Suzuki, H.; et al. Activation of the NRF2 pathway and its impact on the prognosis of anaplastic glioma patients. Neuro-Oncol. 2015, 17, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoll, M.; Lastra, D.; Pajares, M.; Robledinos-Antón, N.; Rojo, A.I.; Fernández-Ginés, R.; Mendiola, M.; Martínez-Marín, V.; Esteban, I.; López-Larrubia, P.; et al. Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biol. 2020, 30, 101425. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, L.A.; Squatrito, M.; Northcott, P.; Awan, A.; Holland, E.C.; Taylor, M.D.; Nahlé, Z.; Kenney, A.M. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene 2012, 31, 1923–1937. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.S.; Kahl, S.B.; Kaye, A.H.; Stylli, S.S.; Koo, M.S.; Gonzales, M.F.; Vardaxis, N.J.; Johnson, C.I. Selective tumor uptake of a boronated porphyrin in an animal model of cerebral glioma. Proc. Natl. Acad. Sci. USA 1992, 89, 1785–1789. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.S.; Kaye, A.H.; Sawyer, W.H.; Morstyn, G.; Megison, P.D.; Stylli, S.S. Selective uptake of hematoporphyrin derivative into human cerebral glioma. Neurosurgery 1990, 26, 248–254. [Google Scholar] [CrossRef]
- Gibault, F.; Bailly, F.; Corvaisier, M.; Coevoet, M.; Huet, G.; Melnyk, P.; Cotelle, P. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem 2017, 12, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Guan, W. Valproic Acid: A Promising Therapeutic Agent in Glioma Treatment. Front. Oncol. 2021, 11, 687362. [Google Scholar] [CrossRef]
- Riva, G.; Cilibrasi, C.; Bazzoni, R.; Cadamuro, M.; Negroni, C.; Butta, V.; Strazzabosco, M.; Dalprà, L.; Lavitrano, M.; Bentivegna, A. Valproic Acid Inhibits Proliferation and Reduces Invasiveness in Glioma Stem Cells Through Wnt/beta Catenin Signalling Activation. Genes 2018, 9, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tsai, Y.H.; Tseng, S.H. Valproic Acid Affected the Survival and Invasiveness of Human Glioma Cells Through Diverse Mechanisms. J. Neuro-Oncol. 2012, 109, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Yuan, X.; Hu, Z.; Li, H.; Wu, M.; Yuan, J.; Zhao, Z.; Su, J.; Wang, X.; et al. Valproic Acid Promotes Human Glioma U87 Cells Apoptosis and Inhibits Glycogen Synthase Kinase3beta Through ERK/Akt Signaling. Cell. Physiol. Biochem. 2016, 39, 2173–2185. [Google Scholar] [CrossRef]
- Knüpfer, M.M.; Hernáiz-Driever, P.; Poppenborg, H.; Wolff, J.E.; Cinatl, J. Valproic acid inhibits proliferation and changes expression of CD44 and CD56 of malignant glioma cells in vitro. Anticancer Res. 1998, 18, 3585–3589. [Google Scholar] [PubMed]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 Attenuates Activation of the Hippo Signaling Pathway and Is a Prime Therapeutic Target for Glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wei, Y.; Zhang, L.; Yee, P.P.; Johnson, M.; Zhang, X.; Gulley, M.; Atkinson, J.M.; Trebak, M.; Wang, H.G.; et al. Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ. Oncogene 2019, 38, 120–139. [Google Scholar] [CrossRef]
- Guan, H.; Zhang, H.; Cai, J.; Wu, J.; Yuan, J.; Li, J.; Huang, Z.; Li, M. IKBKE is over-expressed in glioma and contributes to resistance of glioma cells to apoptosis via activating NF-κB. J. Pathol. 2011, 223, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, J.; Zhang, Z.; Zhu, L.; Dong, S.; Guo, G.; Li, R.; Nan, Y.; Yu, K.; Zhong, Y.; et al. Amlexanox, a selective inhibitor of IKBKE, generates anti-tumoral effects by disrupting the Hippo pathway in human glioblastoma cell lines. Cell Death Dis. 2017, 8, e3022. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Zhao, P.; Li, H.; Fu, H.; Liu, X.; Liu, Y.; Wu, J.; Fu, W. Bazedoxifene enhances paclitaxel efficacy to suppress glioblastoma via altering Hippo/YAP pathway. J. Cancer 2020, 11, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Kong, J.; Wang, D.; Lien, L.L.M.; Lien, E.J.C. A survey of Chinese herbal ingredients with liver protection activities. Chin. Med. 2007, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Saller, R.; Meier, R.; Brignoli, R. The use of silymarin in the treatment of liver diseases. Drugs 2001, 61, 2035–2063. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Wei, X.F.; Jiang, Y.Y.; Huang, H.; Yang, Y.; Fan, S.M.; Zang, L.H.; Tashiro, S.I.; Onodera, O.; Ikejima, T. Silibinin induces the generation of nitric oxide in human breast cancerMCF-7 cells. Free Radic. Res. 2010, 44, 577–584. [Google Scholar] [CrossRef]
- Singh, R.P.; Agarwal, R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur. J. Cancer 2005, 41, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.K.; Agarwal, C.; Chan, D.C.; Agarwal, R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol. Rep. 2004, 11, 493–499. [Google Scholar] [CrossRef]
- Kumar, S.; Raina, K.; Agarwal, C.; Agarwal, R. Silibinin strongly inhibits the growth kinetics of colon cancer stem cell enriched spheroids by modulating interleukin 4/6-mediated survival signals. Oncotarget 2014, 5, 4972–4989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momeny, M.; Malehmir, M.; Zakidizaji, M.; Ghasemi, R.; Ghadimi, H.; Shokrgozar, M.A.; Emami, A.H.; Nafissi, S.; Ghavamzadeh, A.; Ghaffari, S.H. Silibinin inhibits invasive properties of human glioblastoma U87MG cells through suppression of cathepsin B and nuclear factor kappa B-mediated induction of matrix metalloproteinase. Anticancer Drugs 2010, 21, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Bai, Z.L.; Tay, V.; Guo, S.Z.; Ren, J.; Shu, M.G. Silibinin Induced Human Glioblastoma Cell Apoptosis Concomitant with Autophagy through Simultaneous Inhibition of mTOR and YAP. Biomed. Res. Int. 2018, 2018, 6165192. [Google Scholar] [CrossRef]
- Jia, M.; Wang, Y.; Guo, Y.; Yu, P.; Sun, Y.; Song, Y.; Zhao, L. Nitidine chloride suppresses epithelial-mesenchymal transition and stem cell-like properties in glioblastoma by regulating JAK2/STAT3 signaling. Cancer Med. 2021, 10, 3113–3128. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Qi, Q.; Huang, B.; Chen, A.; Li, X.; Wang, J. Nitidine chloride inhibits the malignant behavior of human glioblastoma cells by targeting the PI3K/AKT/mTOR signaling pathway. Oncol. Rep. 2016, 36, 2160–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wu, L.; Lian, C.; Lian, S.; Bao, S.; Zhang, J.; Wang, P.; Ma, J.; Li, Y. Nitidine chloride possesses anticancer property in lung cancer cells through activating Hippo signaling pathway. Cell Death Discov. 2020, 6, 91. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GBM CHARACTERIZATION | |
|---|---|
| Classification |
|
| Prognosis | Less than 5% of patients survive more than five years after diagnosis |
| Standard therapy | Radiotherapy followed by chemotherapy with TMZ |
| MDR |
|
| CHEMORESISTANCE MECHANISMS | |
|---|---|
| BBB |
|
| Altered DNA Repair DDR MMR | They confer chemoresistance phenotype to GBM cells |
| Aberran expression of microRNA (miRNA) | miRNA targets are drug transporter genes, proteins involved in ABCB1/P-gp mediated chemoresistance and genes involved in DNA repair mechanisms |
| Exosome release |
|
| PHARMACOLOGICAL OPPORTUNITIES | |
|---|---|
| VP |
|
| VPA |
|
| Amlodipine | Let an increase of cytosolic Ca2+ level which provokes an actin cytoskeleton remodeling. The new assembly of F-actin is capable to start kinase cascade and phosphorylate YAP which will degrade |
| ALX |
|
| BZA | Act as GP130 inhibitor by competing with IL-6 or IL-11 for the interaction of GP130, leading to the deactivation of IL-6/GP130 signaling and accelerating YAP phosphorylation |
| Silibinin | Induce a concentration-dependent downregulation of YAP |
| NC | Increase YAP phosphorylation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casati, G.; Giunti, L.; Iorio, A.L.; Marturano, A.; Galli, L.; Sardi, I. Hippo Pathway in Regulating Drug Resistance of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 13431. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413431
Casati G, Giunti L, Iorio AL, Marturano A, Galli L, Sardi I. Hippo Pathway in Regulating Drug Resistance of Glioblastoma. International Journal of Molecular Sciences. 2021; 22(24):13431. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413431
Chicago/Turabian StyleCasati, Giacomo, Laura Giunti, Anna Lisa Iorio, Arianna Marturano, Luisa Galli, and Iacopo Sardi. 2021. "Hippo Pathway in Regulating Drug Resistance of Glioblastoma" International Journal of Molecular Sciences 22, no. 24: 13431. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413431