
Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
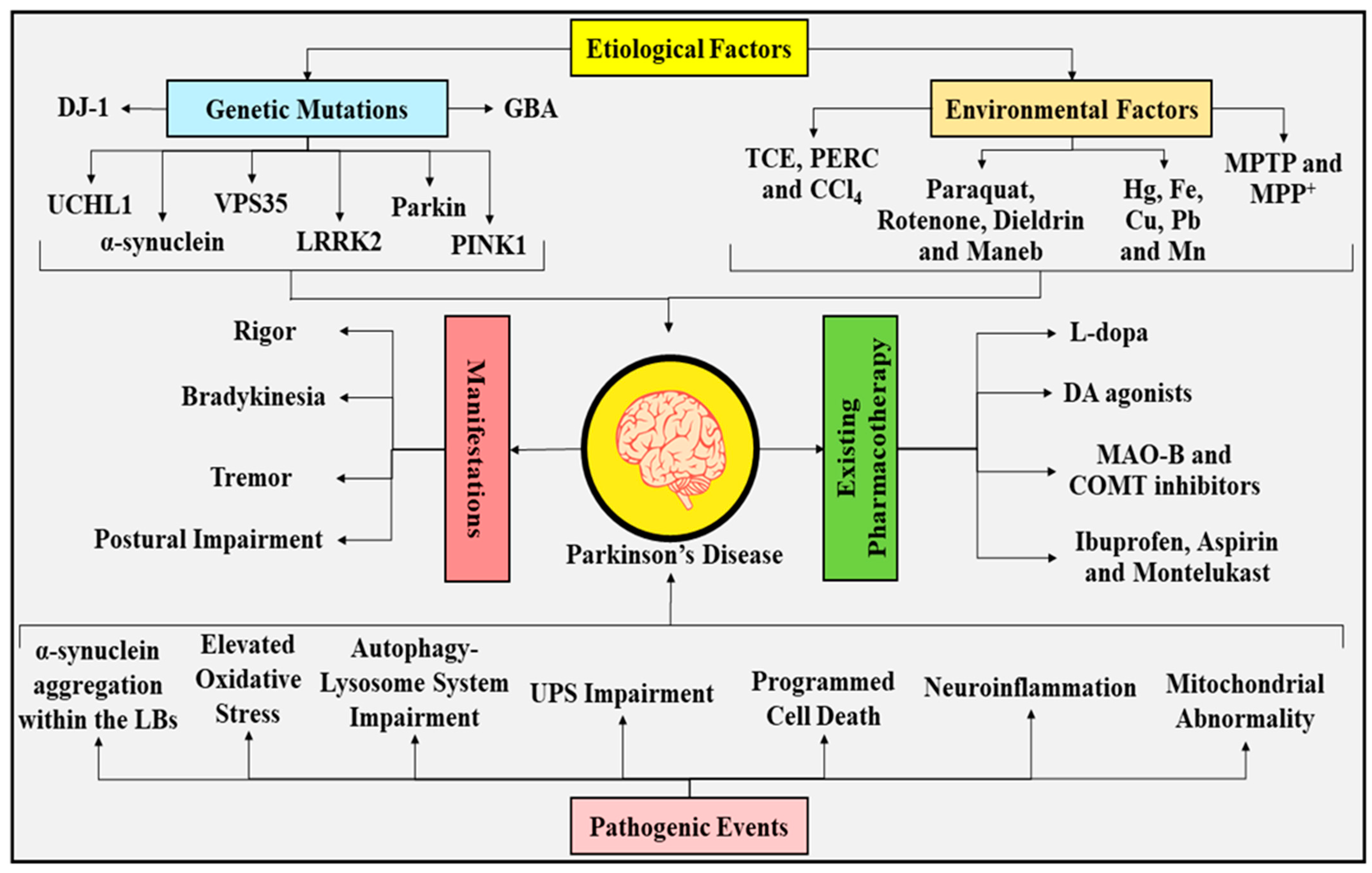
2. Parkinson’s Disease: Etiological Factors, Pathogenic Events, and Treatment
3. Oxidative Stress: An Imperative Player in PD
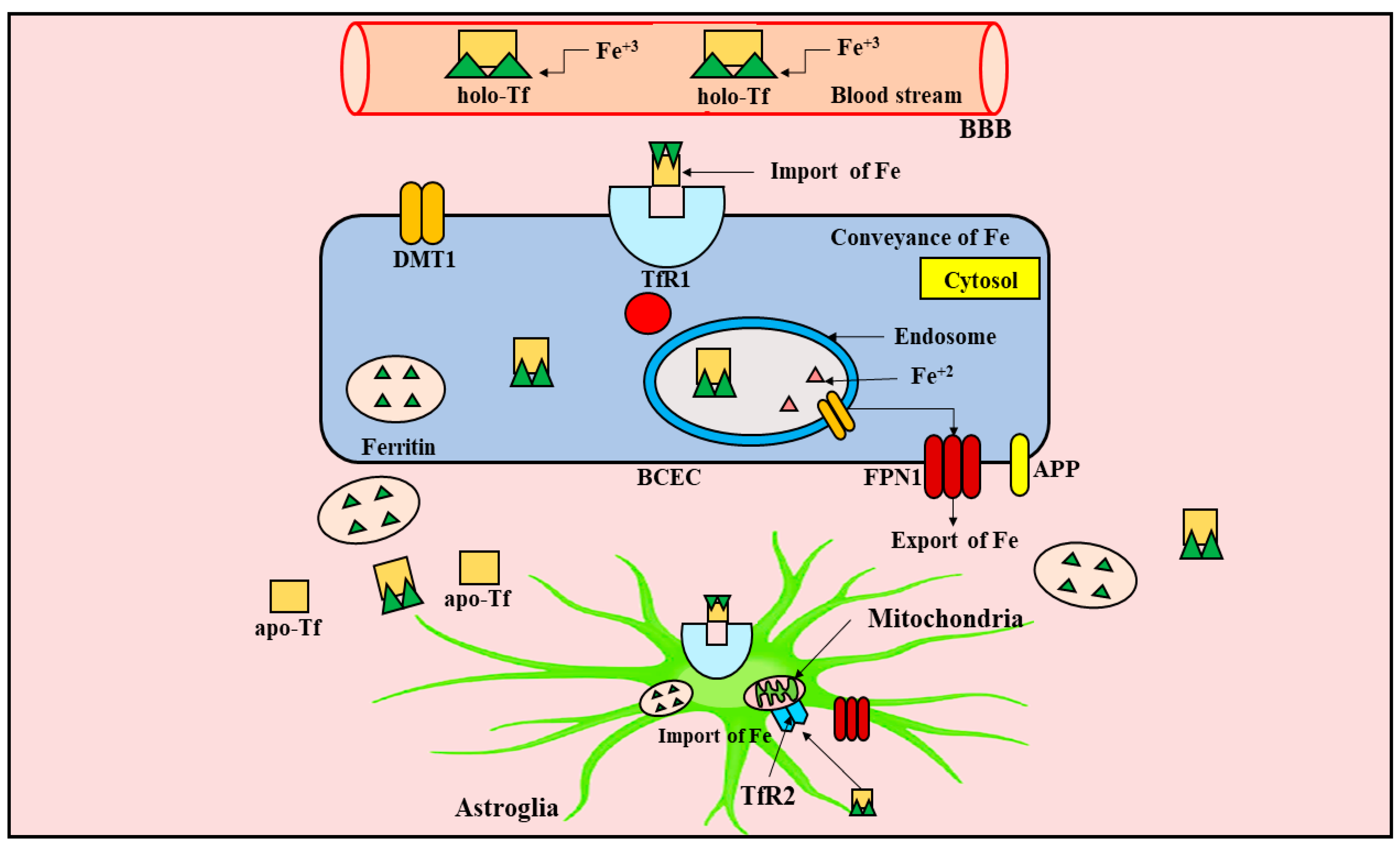
4. Metabolism of Iron in the CNS
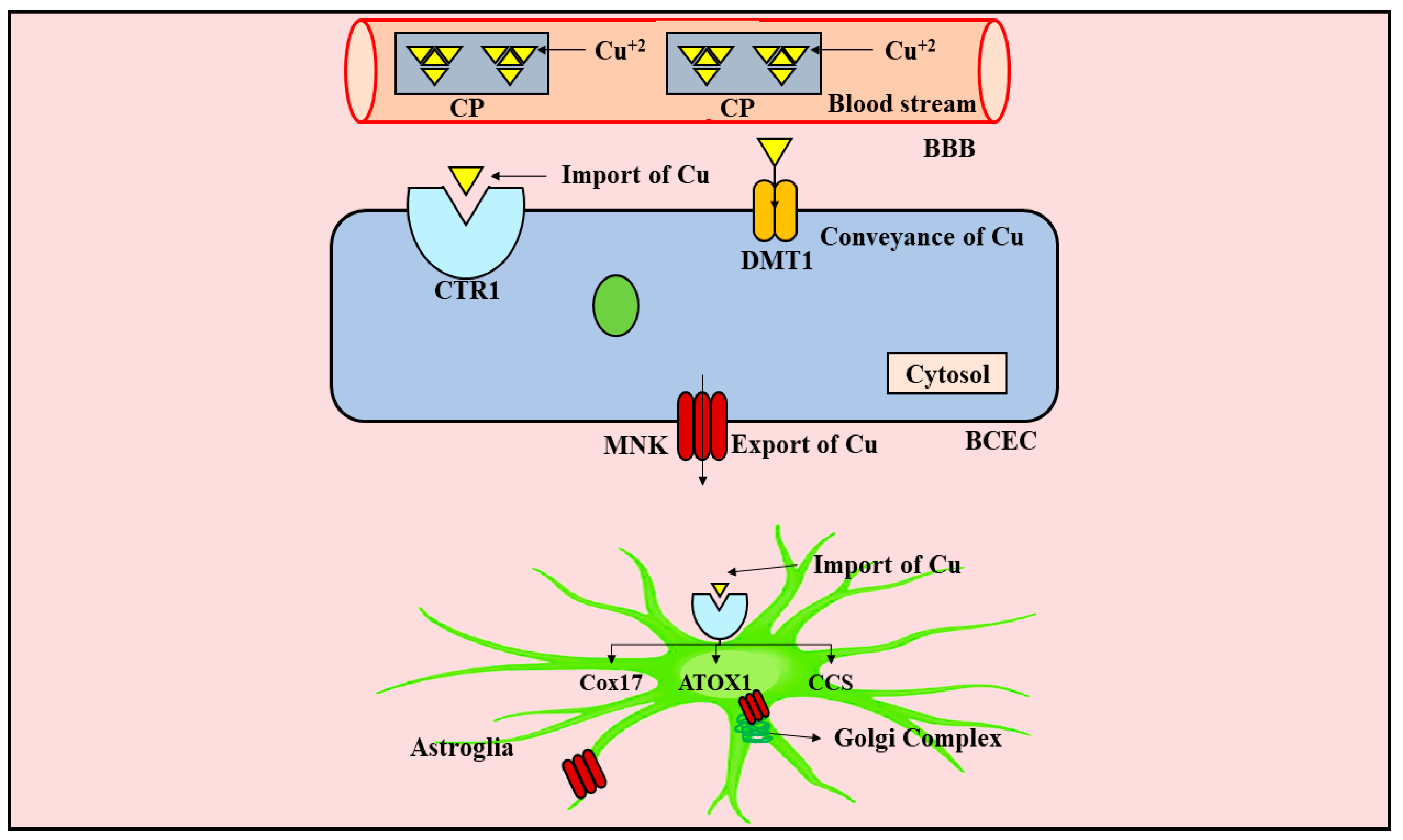
5. Metabolism of Copper in the CNS
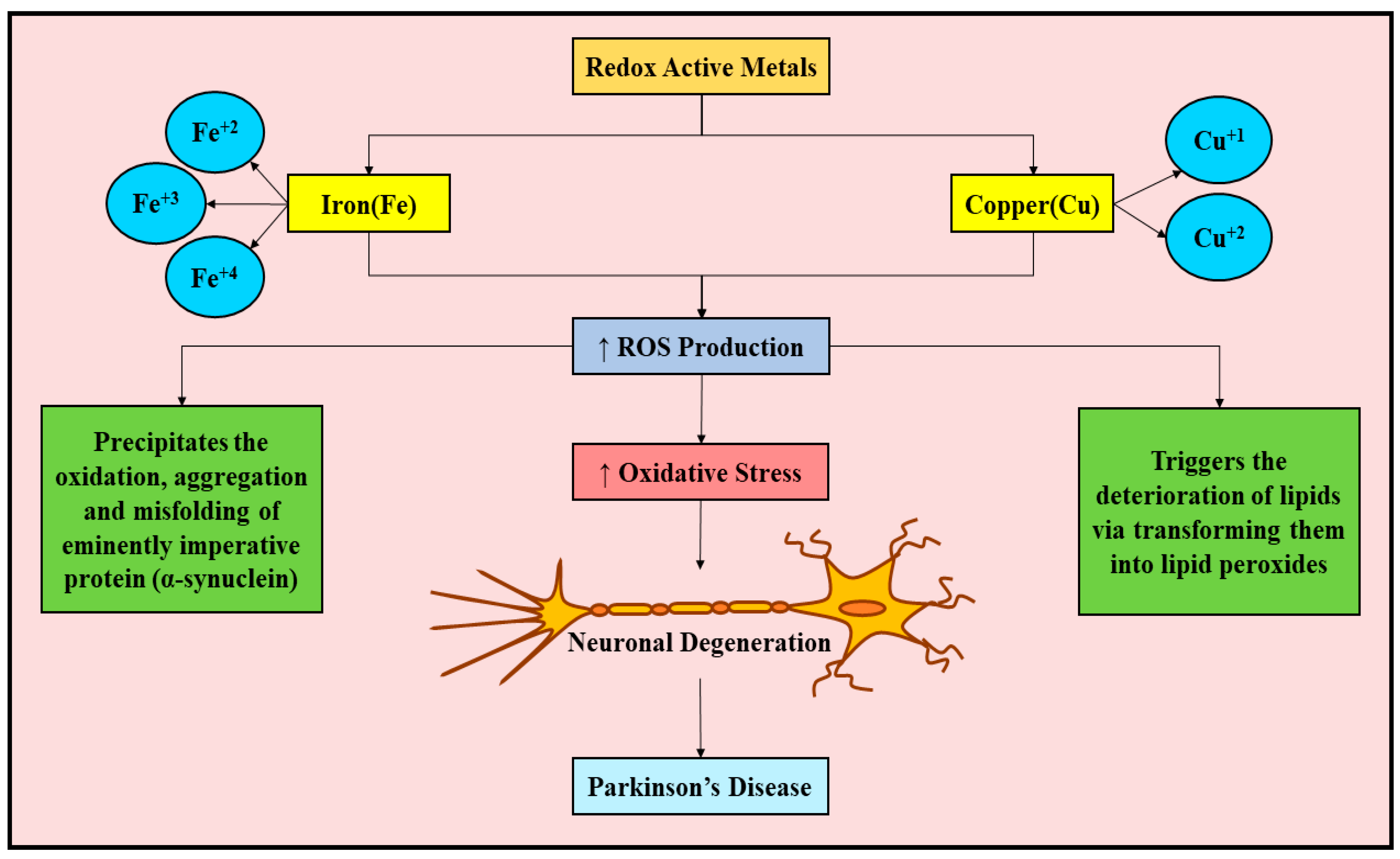
6. Implication and Disrupted Balance of Redox-Active Metals in PD
6.1. Implication of Iron in PD
6.2. Implication of Copper in PD
6.3. Disrupted Balance of Iron and Copper in PD
7. Implication of Metal Complexes and Metal Chelators in the Therapy of PD
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kaur, R.; Mehan, S.; Singh, S. Understanding Multifactorial Architecture of Parkinson’s Disease: Pathophysiology to Management. Neurol. Sci. 2019, 40, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Parkinson’s Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, T.; Terzakis, K. Parkinson Disease. Home Healthc. Now 2016, 34, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Sauerbier, A.; Qamar, M.A.; Rajah, T.; Chaudhuri, K.R. New Concepts in the Pathogenesis and Presentation of Parkinson’s Disease. Clin. Med. 2016, 16, 365. [Google Scholar] [CrossRef] [PubMed]
- Váradi, C. Clinical Features of Parkinson’s Disease: The Evolution of Critical Symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef]
- Surmeier, D.J. Determinants of Dopaminergic Neuron Loss in Parkinson’s Disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Burn, D.J.; Cullen, B.; O’Neill, B.; Evans, J.J.; Coen, R.F.; Lawlor, B.A.; Moxley, R.T.; Laberge, L.; Jean, S.; Richer, L. Journal of Neurology Neurosurgery & Psychiatry. J. Neurol. Neurosurg. Psychiatry 2007, 78, 915. [Google Scholar]
- Reekes, T.H.; Higginson, C.I.; Ledbetter, C.R.; Sathivadivel, N.; Zweig, R.M.; Disbrow, E.A. Sex Specific Cognitive Differences in Parkinson Disease. NPJ Parkinson’s Dis. 2020, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Carola, G. Investigating Early Functional Alteration in a Human IPSC-Based Model of Parkinson’s Disease; Universitat de Barcelona: Barcelona, Spain, 2019. [Google Scholar]
- Shulman, L.M. Gender Differences in Parkinson’s Disease. Gend. Med. 2007, 4, 8–18. [Google Scholar] [CrossRef]
- Picillo, M.; Nicoletti, A.; Fetoni, V.; Garavaglia, B.; Barone, P.; Pellecchia, M.T. The Relevance of Gender in Parkinson’s Disease: A Review. J. Neurol. 2017, 264, 1583–1607. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Przedborski, S. Genetic Clues to the Pathogenesis of Parkinson’s Disease. Nat. Med. 2004, 10, S58–S62. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Vázquez-Vélez, G.E.; Zoghbi, H.Y. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood, N.W. Molecular Pathogenesis of Parkinson’s Disease. Hum. Mol. Genet. 2005, 14, 2749–2755. [Google Scholar] [CrossRef]
- Bonuccelli, U.; del Dotto, P. New Pharmacologic Horizons in the Treatment of Parkinson Disease. Neurology 2006, 67, S30–S38. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Bougea, A.; Piperi, C. Impact of the Apelin/APJ Axis in the Pathogenesis of Parkinson’s Disease with Therapeutic Potential. J. Neurosci. Res. 2021, 99, 2117–2133. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological Treatment of Parkinson Disease: A Review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Dahiya, N. Management of Parkinson׳ s Disease: Current and Future Pharmacotherapy. Eur. J. Pharmacol. 2015, 750, 74–81. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Dulles, VA, USA, 1994; ISBN 0935702725. [Google Scholar]
- Halliwell, B. Oxidative Stress and Neurodegeneration: Where Are We Now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jomova, K.; Baros, S.; Valko, M. Redox Active Metal-Induced Oxidative Stress in Biological Systems. Transit. Met. Chem. 2012, 37, 127–134. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron Neurochemistry in Alzheimer’s Disease and Parkinson’s Disease: Targets for Therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper Signaling in the Mammalian Nervous System: Synaptic Effects. J. NeuroSci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef] [Green Version]
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From Neurotransmission to Neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Angelova, D.M.; Brown, D.R. Iron, Aging, and Neurodegeneration. Metals 2015, 5, 2070–2092. [Google Scholar] [CrossRef] [Green Version]
- Parent, A. A Tribute to James Parkinson. Can. J. Neurol. Sci. 2018, 45, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Zesiewicz, T.A. Parkinson Disease. CONTINUUM Lifelong Learn. Neurol. 2019, 25, 896–918. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Alrashdi, I.; Bungau, S.G. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. Int. J. Mol. Sci. 2021, 22, 10161. [Google Scholar] [CrossRef]
- Rodriguez-Oroz, M.C.; Jahanshahi, M.; Krack, P.; Litvan, I.; Macias, R.; Bezard, E.; Obeso, J.A. Initial Clinical Manifestations of Parkinson’s Disease: Features and Pathophysiological Mechanisms. Lancet Neurol. 2009, 8, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Rampello, L.; Vecchio, I.; Malaguarnera, M.; Battaglia, G.; Rampello, L. Parkinson’s Disease: Therapeutic options. Acta Med. 2012, 28, 117. [Google Scholar]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s Disease: Dopamine, Vesicles and α-Synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S. The Neuropathology of Parkinson’s Disease. Prog. Parkinson Res. 1988, 15, 11–21. [Google Scholar]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Cha, J.; Chung, S.J.; Yoo, H.S.; Sohn, Y.H.; Ye, B.S.; Lee, P.H. Beneficial Effect of Estrogen on Nigrostriatal Dopaminergic Neurons in Drug-Naïve Postmenopausal Parkinson’s Disease. Sci. Rep. 2019, 9, 10531. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.-J.; Lin, C.-H.; Lane, H.-Y. From Menopause to Neurodegeneration—Molecular Basis and Potential Therapy. Int. J. Mol. Sci. 2021, 22, 8654. [Google Scholar] [CrossRef]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s Disease: Mechanisms and Biochemical Processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-Synuclein Structure and Parkinson’s Disease–Lessons and Emerging Principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Piramanayagam, S. Impact of Gene Mutation in the Development of Parkinson’s Disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef]
- Rydning, S.L.; Backe, P.H.; Sousa, M.M.L.; Iqbal, Z.; Oye, A.-M.; Sheng, Y.; Yang, M.; Lin, X.; Slupphaug, G.; Nordenmark, T.H. Novel UCHL1 Mutations Reveal New Insights into Ubiquitin Processing. Hum. Mol. Genet. 2017, 26, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-Q.; Tan, L.; Yu, J.-T. The Role of the LRRK2 Gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.A.; Morrison, B.E. Contributions of VPS35 Mutations to Parkinson’s Disease. Neuroscience 2019, 401, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibánez, P.; Lesage, S.; Lohmann, E.; Thobois, S.; Michele, G.; de Borg, M.; Agid, Y.; Dürr, A.; Brice, A. Mutational Analysis of the PINK1 Gene in Early-Onset Parkinsonism in Europe and North Africa. Brain 2006, 129, 686–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Avenali, M.; Blandini, F.; Cerri, S. Glucocerebrosidase Defects as a Major Risk Factor for Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Samii, A.; Schneer, S.H.; Roberts, J.W.; Griffith, A.; Leis, B.C.; Schellenberg, G.D.; Sidransky, E.; Bird, T.D.; Leverenz, J.B. Glucocerebrosidase Gene Mutations: A Risk Factor for Lewy Body Disorders. Arch. Neurol. 2008, 65, 379–382. [Google Scholar] [CrossRef] [Green Version]
- De Miranda, B.R.; Greenamyre, J.T. Trichloroethylene, a Ubiquitous Environmental Contaminant in the Risk for Parkinson’s Disease. Environ. Sci. Process. Impacts 2020, 22, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M.; Quinlan, P.J.; Ross, G.W.; Marras, C.; Meng, C.; Bhudhikanok, G.S.; Comyns, K.; Korell, M.; Chade, A.R.; Kasten, M. Solvent Exposures and Parkinson Disease Risk in Twins. Ann. Neurol. 2012, 71, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Kochmanski, J.; VanOeveren, S.E.; Patterson, J.R.; Bernstein, A.I. Developmental Dieldrin Exposure Alters DNA Methylation at Genes Related to Dopaminergic Neuron Development and Parkinson’s Disease in Mouse Midbrain. Toxicol. Sci. 2019, 169, 593–607. [Google Scholar] [CrossRef]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s Disease and Residential Exposure to Maneb and Paraquat from Agricultural Applications in the Central Valley of California. Am. J. Epidemiol. 2009, 169, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Mustapha, M.; Taib, C.N.M. MPTP-Induced Mouse Model of Parkinson’s Disease: A Promising Direction for Therapeutic Strategies. Bosn. J. Basic Med Sci. 2021, 21, 422. [Google Scholar]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, P.; Sangar, M.C.; Singh, S.; Tang, W.; Bansal, S.; Chowdhury, G.; Cheng, Q.; Fang, J.-K.; Martin, M.; Guengerich, F.P. Metabolism of 1-Methyl-4-Phenyl-1, 2, 3, 6-Tetrahydropyridine by Mitochondrion-Targeted Cytochrome P450 2D6: Implications in Parkinson Disease. J. Biol. Chem. 2013, 288, 4436–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Xu, J.; Wang, J.; Tong, J.; Bai, X.; Li, H.; Wang, Z.; Huang, Y.; Wu, Y.; Yu, M. Dynamic Changes in the Nigrostriatal Pathway in the MPTP Mouse Model of Parkinson’s Disease. Parkinson’s Dis. 2017, 2017, 9487. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P. Parkinson’s Disease Brain Mitochondrial Complex I Has Oxidatively Damaged Subunits and Is Functionally Impaired and Misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Hartley, A.; Cooper, J.M.; Schapira, A.H. Iron Induced Oxidative Stress and Mitochondrial Dysfunction: Relevance to Parkinson’s Disease. Brain Res. 1993, 627, 349–353. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.; Yokoyama, R.; Shibata, T.; Dawson, V.L.; Dawson, T.M. Role of Neuronal Nitric Oxide in 1-Methyl-4-Phenyl-1, 2, 3, 6-Tetrahydropyridine (MPTP)-Induced Dopaminergic Neurotoxicity. Proc. Natl. Acad. Sci. USA 1996, 93, 4565–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskum, G.; Starkov, A.; Polster, B.M.; Chinopoulos, C. Mitochondrial Mechanisms of Neural Cell Death and Neuroprotective Interventions in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2003, 991, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Matthews, R.T.; Muqit, M.M.K.; Browne, S.E.; Beal, M.F. Inhibition of Neuronal Nitric Oxide Synthase by 7-nitroindazole Protects against MPTP-induced Neurotoxicity in Mice. J. Neurochem. 1995, 64, 936–939. [Google Scholar] [CrossRef]
- Watts, R.N.; Ponka, P.; Richardson, D.R. Effects of Nitrogen Monoxide and Carbon Monoxide on Molecular and Cellular Iron Metabolism: Mirror-Image Effector Molecules That Target Iron. Biochem. J. 2003, 369, 429–440. [Google Scholar] [CrossRef]
- Virarkar, M.; Alappat, L.; Bradford, P.G.; Awad, A.B. L-Arginine and Nitric Oxide in CNS Function and Neurodegenerative Diseases. Crit. Rev. Food Sci. Nutr. 2013, 53, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Fujishiro, H.; Orr, C.; DelleDonne, A.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Parisi, J.E.; Klos, K.J.; Ahlskog, J.E. Neuropathology of Non-Motor Features of Parkinson Disease. Parkinsonism Relat. Disord. 2009, 15, S1–S5. [Google Scholar] [CrossRef]
- Kaur, G.; Behl, T.; Bungau, S.; Kumar, A.; Uddin, M.S.; Mehta, V.; Zengin, G.; Mathew, B.; Shah, M.A.; Arora, S. Dysregulation of the Gut-Brain Axis, Dysbiosis and Influence of Numerous Factors on Gut Microbiota Associated Parkinson’s Disease. Curr. Neuropharmacol. 2021, 19, 233–247. [Google Scholar] [CrossRef]
- Wolters, E.C. Non-Motor Extranigral Signs and Symptoms in Parkinson’s Disease. Parkinsonism Relat. Disord. 2009, 15, S6–S12. [Google Scholar] [CrossRef]
- Ikemura, M.; Saito, Y.; Sengoku, R.; Sakiyama, Y.; Hatsuta, H.; Kanemaru, K.; Sawabe, M.; Arai, T.; Ito, G.; Iwatsubo, T. Lewy Body Pathology Involves Cutaneous Nerves. J. Neuropathol. Exp. Neurol. 2008, 67, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Michell, A.W.; Luheshi, L.M.; Barker, R.A. Skin and Platelet α-Synuclein as Peripheral Biomarkers of Parkinson’s Disease. Neurosci. Lett. 2005, 381, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Shishido, T.; Ikemura, M.; Obi, T.; Yamazaki, K.; Terada, T.; Sugiura, A.; Saito, Y.; Murayama, S.; Mizoguchi, K. α-Synuclein Accumulation in Skin Nerve Fibers Revealed by Skin Biopsy in Pure Autonomic Failure. Neurology 2010, 74, 608–610. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; del Tredici, K. Stages in the Development of Parkinson’s Disease-Related Pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; Deurwaerdère, P. de L-Dopa in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning. Int. J. Mol. Sci. 2020, 21, 294. [Google Scholar] [CrossRef] [Green Version]
- Rinne, J.O.; Portin, R.; Ruottinen, H.; Nurmi, E.; Bergman, J.; Haaparanta, M.; Solin, O. Cognitive Impairment and the Brain Dopaminergic System in Parkinson Disease:[18F] Fluorodopa Positron Emission Tomographic Study. Arch. Neurol. 2000, 57, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Bogetofte, H.; Alamyar, A.; Blaabjerg, M.; Meyer, M. Levodopa Therapy for Parkinson’s Disease: History, Current Status and Perspectives. CNS Neurol. Disord. Drug Targets 2020, 19, 572–583. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: Their Effects on Brain Dopamine Levels and Uses in Parkinson’s Disease. J. Neural Transm. 2019, 126, 433–448. [Google Scholar] [CrossRef]
- Salat, D.; Tolosa, E. Levodopa in the Treatment of Parkinson’s Disease: Current Status and New Developments. J. Parkinson’s Dis. 2013, 3, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zhang, M.; Li, C.; Jiang, X.; Su, Y.; Zhang, Y. Benefits of Vitamins in the Treatment of Parkinson’s Disease. Oxidative Med. Cell. Longev. 2019, 2019, 6867. [Google Scholar] [CrossRef]
- Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. [Google Scholar] [CrossRef]
- Fyfe, I. Aspirin and Ibuprofen Could Lower Risk of LRRK2 Parkinson Disease. Nat. Rev. Neurol. 2020, 16, 460. [Google Scholar] [CrossRef]
- Domínguez-Baleón, C.; Ong, J.-S.; Scherzer, C.R.; Rentería, M.E.; Dong, X. Understanding the Effect of Smoking and Drinking Behavior on Parkinson’s Disease Risk: A Mendelian Randomization Study. Sci. Rep. 2021, 11, 13980. [Google Scholar]
- Ren, X.; Chen, J.-F. Caffeine and Parkinson’s Disease: Multiple Benefits and Emerging Mechanisms. Front. Neurosci. 2020, 14, 1334. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. Oxidative Stress: Biomarkers and Novel Therapeutic Pathways. Exp. Gerontol. 2010, 45, 217–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Jiang, H.; Xie, J. Alcohol intake and risk of Parkinson’s disease: A meta-analysis of observational studies. Mov. Disord. 2014, 29, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Yang, Q.; Joshi, R.B.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernan, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, C.B.; Berry, C.; Chang, E.T.; Sielken Jr, R.L.; Mandel, J.S. Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: Systematic review and meta-analysis. PLoS ONE 2016, 11, e0151841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.; Dunaev, A. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Marlatt, M.; Lee, H.; Perry, G.; Smith, M.A.; Zhu, X. Sources and Mechanisms of Cytoplasmic Oxidative Damage in Alzheimer’s Disease. Acta Neurobiol. Exp. 2004, 64, 81–88. [Google Scholar]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative Diseases and Oxidative Stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Crabtree, D.M.; Zhang, J. Genetically Engineered Mouse Models of Parkinson’s Disease. Brain Res. Bull. 2012, 88, 13–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.A.; Smith, R.A.; Safe, S.; Szabo, C.; Tjalkens, R.B.; Robertson, F.M. Toxicological and Pathophysiological Roles of Reactive Oxygen and Nitrogen Species. Toxicology 2010, 276, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Mytilineou, C.; Kramer, B.C.; Yabut, J.A. Glutathione Depletion and Oxidative Stress. Parkinsonism Relat. Disord. 2002, 8, 385–387. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Jia, J.-J.; Kwon, Y.; Wang, S.-D.; Bai, J. The Role of Thioredoxin-1 in Suppression of Endoplasmic Reticulum Stress in Parkinson Disease. Free. Radic. Biol. Med. 2014, 67, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, G.; Szeto, S.S.W.; Chong, C.M.; Quan, Q.; Huang, C.; Cui, W.; Guo, B.; Wang, Y.; Han, Y. Examining the Neuroprotective Effects of Protocatechuic Acid and Chrysin on in Vitro and in Vivo Models of Parkinson Disease. Free. Radic. Biol. Med. 2015, 84, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Olanow, C.W. The Pathogenesis of Cell Death in Parkinson’s Disease. Neurology 2006, 66, S24–S36. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, S.-C.; Chen, K.-Y.; Lai, J.-H.; Wu, C.-C.; Yu, Y.-W.; Luo, Y.; Hsieh, T.-H.; Chiang, Y.-H. Voluntary Physical Exercise Improves Subsequent Motor and Cognitive Impairments in a Rat Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Yin, J.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Huang, S. Hydrogen Peroxide Inhibits MTOR Signaling by Activation of AMPK α Leading to Apoptosis of Neuronal Cells. Lab. Investig. 2010, 90, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S. Rotenone Induction of Hydrogen Peroxide Inhibits MTOR-Mediated S6K1 and 4E-BP1/EIF4E Pathways, Leading to Neuronal Apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojha, S.; Javed, H.; Azimullah, S.; Khair, S.B.A.; Haque, M.E. Glycyrrhizic Acid Attenuates Neuroinflammation and Oxidative Stress in Rotenone Model of Parkinson’s Disease. Neurotox. Res. 2016, 29, 275–287. [Google Scholar] [CrossRef]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.-F.; Benabid, A.-L.; Sadoul, R.; Verna, J.-M. Molecular Pathways Involved in the Neurotoxicity of 6-OHDA, Dopamine and MPTP: Contribution to the Apoptotic Theory in Parkinson’s Disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Mythri, R.B.; Venkateshappa, C.; Harish, G.; Mahadevan, A.; Muthane, U.B.; Yasha, T.C.; Bharath, M.M.S.; Shankar, S.K. Evaluation of Markers of Oxidative Stress, Antioxidant Function and Astrocytic Proliferation in the Striatum and Frontal Cortex of Parkinson’s Disease Brains. Neurochem. Res. 2011, 36, 1452–1463. [Google Scholar] [CrossRef]
- Vinish, M.; Anand, A.; Prabhakar, S. Altered Oxidative Stress Levels in Indian Parkinson’s Disease Patients with PARK2 Mutations. Acta Biochim. Pol. 2011, 58, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Buhmann, C.; Arlt, S.; Kontush, A.; Moller-Bertram, T.; Sperber, S.; Oechsner, M.; Stuerenburg, H.-J.; Beisiegel, U. Plasma and CSF Markers of Oxidative Stress Are Increased in Parkinson’s Disease and Influenced by Antiparkinsonian Medication. Neurobiol. Dis. 2004, 15, 160–170. [Google Scholar] [CrossRef]
- Quilty, M.C.; King, A.E.; Gai, W.P.; Pountney, D.L.; West, A.K.; Vickers, J.C.; Dickson, T.C. Alpha-Synuclein Is Upregulated in Neurones in Response to Chronic Oxidative Stress and Is Associated with Neuroprotection. Exp. Neurol. 2006, 199, 249–256. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hsu, L.J.; Xia, Y.; Takeda, A.; Sisk, A.; Sundsmo, M.; Masliah, E. Oxidative Stress Induces Amyloid-like Aggregate Formation of NACP/α-Synuclein in Vitro. Neuroreport 1999, 10, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.-Y.; Ischiropoulos, H. Dityrosine Cross-Linking Promotes Formation of Stable α-Synuclein Polymers: Implication of Nitrative and Oxidative Stress in the Pathogenesis of Neurodegenerative Synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Zhao, X.; Li, Y.; Li, G.; Liu, X. Damage to Dopaminergic Neurons by Oxidative Stress in Parkinson’s Disease. Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.K. Oxidative Stress in Neurodegeneration: Cause or Consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Rouault, T.A. Iron Metabolism in the CNS: Implications for Neurodegenerative Diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a Ferrireductase Required for Efficient Transferrin-Dependent Iron Uptake in Erythroid Cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moos, T.; Morgan, E.H. The Significance of the Mutated Divalent Metal Transporter (DMT1) on Iron Transport into the Belgrade Rat Brain. J. Neurochem. 2004, 88, 233–245. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 Is an Iron and Zinc Transporter Whose Cell-Surface Expression is Up-Regulated by Cellular Iron Loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [Green Version]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 Is a Complex Broad-Scope Metal-Ion Transporter Whose Functional Properties Support Roles in the Cellular Uptake of Zinc and Nontransferrin-Bound Iron. Am. J. Physiol.-Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [Green Version]
- Van Raaij, S.E.G.; Srai, S.K.S.; Swinkels, D.W.; van Swelm, R.P.L. Iron Uptake by ZIP8 and ZIP14 in Human Proximal Tubular Epithelial Cells. Biometals 2019, 32, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like Protein 14 (ZIP14) Promotes the Cellular Assimilation of Iron from Transferrin. J. Biol. Chem. 2010, 285, 32141–32150. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Kosman, D.J. Molecular Mechanisms of Non-transferrin-bound and Transferring-bound Iron Uptake in Primary Hippocampal Neurons. J. Neurochem. 2015, 133, 668–683. [Google Scholar] [CrossRef] [Green Version]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.-S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H. Iron Chaperones PCBP1 and PCBP2 Mediate the Metallation of the Dinuclear Iron Enzyme Deoxyhypusine Hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef] [Green Version]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the HIF Prolyl Hydroxylase by the Iron Chaperones PCBP1 and PCBP2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A. Each Member of the Poly-r (C)-Binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantopoulos, K. Iron Metabolism and the IRE/IRP Regulatory System: An Update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The Physiological Functions of Iron Regulatory Proteins in Iron Homeostasis-an Update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Gerlach, M.; Schünemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.H.; Riederer, P.; Ben-Shachar, D. Iron-Binding Characteristics of Neuromelanin of the Human Substantia Nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in Iron-Depleted and Iron-Replete Cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheykhansari, S.; Kozielski, K.; Bill, J.; Sitti, M.; Gemmati, D.; Zamboni, P.; Singh, A.V. Redox Metals Homeostasis in Multiple Sclerosis and Amyotrophic Lateral Sclerosis: A Review. Cell Death Dis. 2018, 9, 348. [Google Scholar] [CrossRef]
- Choi, B.-S.; Zheng, W. Copper Transport to the Brain by the Blood-Brain Barrier and Blood-CSF Barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.A.; Durley, A.P.; Prohaska, J.R.; Harris, Z.L. Copper Transport and Metabolism Are Normal in Aceruloplasminemic Mice. J. Biol. Chem. 2001, 276, 36857–36861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular Glutathione Plays a Key Role in Copper Uptake Mediated by Human Copper Transporter 1. Am. J. Physiol. Cell Physiol. 2013, 304, C768–C779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia, L.; González-Agüero, M.; Cisternas, M.F.; Suazo, M.; Cambiazo, V.; Uauy, R.; González, M. Metallothionein Is Crucial for Safe Intracellular Copper Storage and Cell Survival at Normal and Supra-Physiological Exposure Levels. Biochem. J. 2004, 378, 617–624. [Google Scholar] [CrossRef] [Green Version]
- La Fontaine, S.; Mercer, J.F.B. Trafficking of the Copper-ATPases, ATP7A and ATP7B: Role in Copper Homeostasis. Arch. Biochem. Biophys. 2007, 463, 149–167. [Google Scholar] [CrossRef]
- Linz, R.; Lutsenko, S. Copper-Transporting ATPases ATP7A and ATP7B: Cousins, Not Twins. J. Bioenerg. Biomembr. 2007, 39, 403–407. [Google Scholar] [CrossRef]
- Greenough, M.; Pase, L.; Voskoboinik, I.; Petris, M.J.; O’Brien, A.W.; Camakaris, J. Signals Regulating Trafficking of Menkes (MNK.; ATP7A) Copper-Translocating P-Type ATPase in Polarized MDCK Cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1463–C1471. [Google Scholar] [CrossRef]
- Pase, L.; Voskoboinik, I.; Greenough, M.; Camakaris, J. Copper Stimulates Trafficking of a Distinct Pool of the Menkes Copper ATPase (ATP7A) to the Plasma Membrane and Diverts It into a Rapid Recycling Pool. Biochem. J. 2004, 378, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Brunette, K.E.; Tran, P.V.; Wobken, J.D.; Carlson, E.S.; Georgieff, M.K. Gestational and Neonatal Iron Deficiency Alters Apical Dendrite Structure of CA1 Pyramidal Neurons in Adult Rat Hippocampus. Dev. Neurosci. 2010, 32, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhukovskaya, E.; Karelin, A.; Rumyantsev, A. Neurocognitive Dysfunctions in Iron Deficiency Patients; IntechOpen: London, UK, 2019; pp. 83–113. [Google Scholar]
- Garza-Lombo, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, M.; Puig, N.; Carbonell, T.; Castillo, J.; Martinez, J.M.; Rama, R.; Dávalos, A. Iron Intake Increases Infarct Volume after Permanent Middle Cerebral Artery Occlusion in Rats. Brain Res. 2002, 952, 1–6. [Google Scholar] [CrossRef]
- Cheah, J.H.; Kim, S.F.; Hester, L.D.; Clancy, K.W.; Patterson III, S.E.; Papadopoulos, V.; Snyder, S.H. NMDA Receptor-Nitric Oxide Transmission Mediates Neuronal Iron Homeostasis via the GTPase Dexras1. Neuron 2006, 51, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Halliday, G.M.; Henderson, J.; Griffiths, F.M.; Heinemann, T.; Riederer, P.; Gerlach, M. The Dopamine Receptor Agonist Lisuride Attenuates Iron-Mediated Dopaminergic Neurodegeneration. Exp. Neurol. 2003, 184, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Double, K.L.; Youdim, M.B.H.; Riederer, P. Potential Sources of Increased Iron in the Substantia Nigra of Parkinsonian Patients. Parkinson’s Dis. Relat. Disord. 2006, 9, 133–142. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The Relevance of Iron in the Pathogenesis of Parkinson’s Disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Youdim, M.B.; Riederer, P. The Role of Iron in Senescence of Dopaminergic Neurons in Parkinson’s Disease. J. Neural Transm. 1993, 40, 57–67. [Google Scholar]
- Riederer, P.; Sofic, E.; Rausch, W.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B.H. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Shoham, S.; Youdim, M.B.H. The Effects of Iron Deficiency and Iron and Zinc Supplementation on Rat Hippocampus Ferritin. J. Neural Transm. 2002, 109, 1241–1256. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and Aluminum Increase in the Substantia Nigra of Patients with Parkinson’s Disease: An X-ray Microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef]
- Berg, D.; Grote, C.; Rausch, W.-D.; Mäurer, M.; Wesemann, W.; Riederer, P.; Becker, G. Iron Accumulation in the Substantia Nigra in Rats Visualized by Ultrasound. Ultrasound Med. Biol. 1999, 25, 901–904. [Google Scholar] [CrossRef]
- Gorell, J.M.; Ordidge, R.J.; Brown, G.G.; Deniau, J.C.; Buderer, N.M.; Helpern, J.A. Increased Iron-related MRI Contrast in the Substantia Nigra in Parkinson’s Disease. Neurology 1995, 45, 1138–1143. [Google Scholar] [CrossRef]
- Jellinger, K.; Kienzl, E.; Rumpelmair, G.; Riederer, P.; Stachelberger, H.; Ben-Shachar, D.; Youdim, M.B.H. Iron-melanin Complex in Substantia Nigra of Parkinsonian Brains: An X-ray Microanalysis. J. Neurochem. 1992, 59, 1168–1171. [Google Scholar] [CrossRef]
- Mastroberardino, P.G.; Hoffman, E.K.; Horowitz, M.P.; Betarbet, R.; Taylor, G.; Cheng, D.; Na, H.M.; Gutekunst, C.-A.; Gearing, M.; Trojanowski, J.Q. A Novel Transferrin/TfR2-Mediated Mitochondrial Iron Transport System Is Disrupted in Parkinson’s Disease. Neurobiol. Dis. 2009, 34, 417–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, M.; Double, K.L.; Youdim, M.B.H.; Riederer, P. Strategies for the Protection of Dopaminergic Neurons against Neurotoxicity. Neurotox. Res. 2000, 2, 99–114. [Google Scholar] [CrossRef]
- Zhu, W.; Li, X.; Xie, W.; Luo, F.; Kaur, D.; Andersen, J.K.; Jankovic, J.; Le, W. Genetic Iron Chelation Protects against Proteasome Inhibition-Induced Dopamine Neuron Degeneration. Neurobiol. Dis. 2010, 37, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.T.; Gallardo, V.; Muñoz, P.; Tapia, V.; Esparza, A.; Salazar, J.; Speisky, H. Progressive Iron Accumulation Induces a Biphasic Change in the Glutathione Content of Neuroblastoma Cells. Free. Radic. Biol. Med. 2004, 37, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular Iron Transport and Storage: From Molecular Mechanisms to Health Implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H. The Haber-Weiss Cycle–70 Years Later. Redox Rep. 2001, 6, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Jiang, D.H. Excessive Iron Accumulation in the Brain: A Possible Potential Risk of Neurodegeneration in Parkinson’s Disease. J. Neural Transm. 1997, 104, 649–660. [Google Scholar] [CrossRef]
- Lan, A.P.; Chen, J.; Chai, Z.F.; Hu, Y. The Neurotoxicity of Iron, Copper and Cobalt in Parkinson’s Disease through ROS-Mediated Mechanisms. Biometals 2016, 29, 665–678. [Google Scholar] [CrossRef]
- Sangchot, P.; Sharma, S.; Chetsawang, B.; Porter, J.; Govitrapong, P.; Ebadi, M. Deferoxamine Attenuates Iron-Induced Oxidative Stress and Prevents Mitochondrial Aggregation and α-Synuclein Translocation in SK-N-SH Cells in Culture. Dev. Neurosci. 2002, 24, 143–153. [Google Scholar] [CrossRef]
- Li, W.-J.; Jiang, H.; Song, N.; Xie, J.-X. Dose-and Time-Dependent α-Synuclein Aggregation Induced by Ferric Iron in SK-N-SH Cells. Neurosci. Bull. 2010, 26, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Jiang, H.; Song, N.; Xie, J. Oxidative Stress Partially Contributes to Iron-Induced Alpha-Synuclein Aggregation in SK-N-SH Cells. Neurotox. Res. 2011, 19, 435–442. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Song, N.; Xu, H.; Wang, R.; Xie, J.; Jiang, H. Alpha-Synuclein Aggregation Is Involved in the Toxicity Induced by Ferric Iron to SK-N-SH Neuroblastoma Cells. J. Neural Transm. 2011, 118, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Högen, T.; Hillmer, A.S.; Bader, B.; Schmidt, F.; Kamp, F.; Kretzschmar, H.A.; Botzel, K.; Giese, A. Generation of Ferric Iron Links Oxidative Stress to α-Synuclein Oligomer Formation. J. Parkinson’s Dis. 2011, 1, 205–216. [Google Scholar] [CrossRef] [PubMed]
- George, J.L.; Mok, S.; Moses, D.; Wilkins, S.; Bush, A.I.; Cherny, R.A.; Finkelstein, D.I. Targeting the Progression of Parkinson’s Disease. Curr. Neuropharmacol. 2009, 7, 9–36. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the Acidic C-Terminal Region Comprising Amino Acids 109− 140 on α-Synuclein Aggregation in Vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef]
- Gomez, F.J.; Aguirre, P.; Gonzalez-Billault, C.; Núñez, M.T. Iron Mediates Neuritic Tree Collapse in Mesencephalic Neurons Treated with 1-Methyl-4-Phenylpyridinium (MPP+). J. Neural Transm. 2011, 118, 421–431. [Google Scholar] [CrossRef]
- Acevedo, K.; Masaldan, S.; Opazo, C.M.; Bush, A.I. Redox Active Metals in Neurodegenerative Diseases. JBIC J. Biol. Inorg. Chem. 2019, 24, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra during Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Cardenas, S.; Huenchuguala, S.; Briceño, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Alpha-Synuclein Oligomer Formation and Neurotoxicity. Toxicol. Sci. 2015, 145, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Maker, H.S.; Weiss, C.; Silides, D.J.; Cohen, G. Coupling of Dopamine Oxidation (Monoamine Oxidase Activity) to Glutathione Oxidation via the Generation of Hydrogen Peroxide in Rat Brain Homogenates. J. Neurochem. 1981, 36, 589–593. [Google Scholar] [CrossRef]
- McCance, R.A.; Widdowson, E.M. The Absorption and Excretion of Iron Following Oral and Intravenous Administration. J. Physiol. 1938, 94, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shachar, D.; Eshel, G.; Finberg, J.P.M.; Youdim, M.B.H. The Iron Chelator Desferrioxamine (Desferal) Retards 6-hydroxydopamine-induced Degeneration of Nigrostriatal Dopamine Neurons. J. Neurochem. 1991, 56, 1441–1444. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Gaeta, A.; Francis, P.T.; Williams, R.J.; Hider, R.C. Neuroprotective Actions of Deferiprone in Cultured Cortical Neurones and SHSY-5Y Cells. J. Neurochem. 2008, 105, 2466–2476. [Google Scholar] [CrossRef]
- Hider, R.C. Potential Protection from Toxicity by Oral Iron Chelators. Toxicol. Lett. 1995, 82, 961–967. [Google Scholar] [CrossRef]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically Available Iron Chelators Induce Neuroprotection in the 6-OHDA Model of Parkinson’s Disease after Peripheral Administration. J. Neural Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals Associated Neurodegeneration in Parkinson’s Disease: Insight to Physiological, Pathological Mechanisms and Management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef]
- Davies, K.M.; Mercer, J.F.B.; Chen, N.; Double, K.L. Copper Dyshomoeostasis in Parkinson’s Disease: Implications for Pathogenesis and Indications for Novel Therapeutics. Clin. Sci. 2016, 130, 565–574. [Google Scholar] [CrossRef]
- Willis, A.W.; Evanoff, B.A.; Lian, M.; Galarza, A.; Wegrzyn, A.; Schootman, M.; Racette, B.A. Metal Emissions and Urban Incident Parkinson Disease: A Community Health Study of Medicare Beneficiaries by Using Geographic Information Systems. Am. J. Epidemiol. 2010, 172, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Tarohda, T.; Ishida, Y.; Kawai, K.; Yamamoto, M.; Amano, R. Regional Distributions of Manganese, Iron, Copper, and Zinc in the Brains of 6-Hydroxydopamine-Induced Parkinsonian Rats. Anal. Bioanal. Chem. 2005, 383, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, D.; Uzun, H. Copper Intoxication; Antioxidant Defenses and Oxidative Damage in Rat Brain. Biol. Trace Elem. Res. 2009, 127, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-R.; Jiang, H.; Wang, J.; Xie, J.-X. Copper (Cu 2+) Induces Degeneration of Dopaminergic Neurons in the Nigrostriatal System of Rats. Neurosci. Bull. 2008, 24, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spisni, E.; Valerii, M.C.; Manerba, M.; Strillacci, A.; Polazzi, E.; Mattia, T.; Griffoni, C.; Tomasi, V. Effect of Copper on Extracellular Levels of Key Pro-Inflammatory Molecules in Hypothalamic GN11 and Primary Neurons. Neurotoxicology 2009, 30, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Uriu-Adams, J.Y.; Scherr, R.E.; Lanoue, L.; Keen, C.L. Influence of Copper on Early Development: Prenatal and Postnatal Considerations. Biofactors 2010, 36, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Kowalik-Jankowska, T.; Rajewska, A.; Jankowska, E.; Grzonka, Z. Products of Cu (II)-Catalyzed Oxidation of α-Synuclein Fragments Containing M 1-D 2 and H 50 Residues in the Presence of Hydrogen Peroxide. Dalton Trans. 2008, 6, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Tavassoly, O.; Nokhrin, S.; Dmitriev, O.Y.; Lee, J.S. Cu (II) and Dopamine Bind to A-synuclein and Cause Large Conformational Changes. FEBS J. 2014, 281, 2738–2753. [Google Scholar] [CrossRef] [Green Version]
- Olanow, C.W.; Brundin, P. Parkinson’s Disease and Alpha Synuclein: Is Parkinson’s Disease a Prion-like Disorder? Mov. Disord. 2013, 28, 31–40. [Google Scholar] [CrossRef]
- Ahmad, A.; Burns, C.S.; Fink, A.L.; Uversky, V.N. Peculiarities of Copper Binding to α-Synuclein. J. Biomol. Struct. Dyn. 2012, 29, 825–842. [Google Scholar] [CrossRef]
- Wright, J.A.; Wang, X.; Brown, D.R. Unique Copper-induced Oligomers Mediate Alpha-synuclein Toxicity. FASEB J. 2009, 23, 2384–2393. [Google Scholar] [CrossRef]
- Wang, X.; Moualla, D.; Wright, J.A.; Brown, D.R. Copper Binding Regulates Intracellular Alpha-synuclein Localisation, Aggregation and Toxicity. J. Neurochem. 2010, 113, 704–714. [Google Scholar] [CrossRef]
- Anandhan, A.; Rodriguez-Rocha, H.; Bohovych, I.; Griggs, A.M.; Zavala-Flores, L.; Reyes-Reyes, E.M.; Seravalli, J.; Stanciu, L.A.; Lee, J.; Rochet, J.-C. Overexpression of Alpha-Synuclein at Non-Toxic Levels Increases Dopaminergic Cell Death Induced by Copper Exposure via Modulation of Protein Degradation Pathways. Neurobiol. Dis. 2015, 81, 76–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowski, H.; Janicka-Klos, A.; Brasun, J.; Gaggelli, E.; Valensin, D.; Valensin, G. Copper, Iron, and Zinc Ions Homeostasis and Their Role in Neurodegenerative Disorders (Metal Uptake, Transport, Distribution and Regulation). Coord. Chem. Rev. 2009, 253, 2665–2685. [Google Scholar] [CrossRef]
- Boll, M.-C.; Sotelo, J.; Otero, E.; Alcaraz-Zubeldia, M.; Rios, C. Reduced Ferroxidase Activity in the Cerebrospinal Fluid from Patients with Parkinson’s Disease. Neurosci. Lett. 1999, 265, 155–158. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-Analysis of Brain Iron Levels of Parkinson’s Disease Patients Determined by Postmortem and MRI Measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar]
- Ayton, S.; Lei, P. Nigral Iron Elevation Is an Invariable Feature of Parkinson’s Disease and Is a Sufficient Cause of Neurodegeneration. BioMed. Res. Int. 2014, 2014, 1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Chen, Y.; Zhang, Y.; Wang, F.; Yu, H.; Zhang, C.; Jiang, Z.; Luo, W. Iron Deposition in Parkinson’s Disease by Quantitative Susceptibility Mapping. BMC Neurosci. 2019, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Azad, M.G.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I. Parkinson’s Disease: Alterations in Iron and Redox Biology as a Key to Unlock Therapeutic Strategies. Redox Biol. 2021, 15, 101896. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content in Postmortem Parkinsonian Brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- Scholefield, M.; Church, S.J.; Xu, J.; Patassini, S.; Roncaroli, F.; Hooper, N.M.; Unwin, R.D.; Cooper, G.J.S. Widespread Decreases in Cerebral Copper Are Common to Parkinson’s Disease Dementia and Alzheimer’s Disease Dementia. Front. Aging Neurosci. 2021, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Rogers, E.; Xia, Y.; Liu, W.; Ma, W.; Zeng, W.; Gong, L.; Fang, J.; et al. The association of iron and the pathologies of Parkinson’s diseases in MPTP/MPP+-induced neuronal degeneration in non-human primates and in cell culture. Front. Aging Neurosci. 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Reyes, S.; Werry, E.L.; Halliday, G.M. Selective Cell Death in Neurodegeneration: Why are some Neurons Spared in Vulnerable Regions? Prog. Neurobiol. 2010, 92, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-W.; Tai, Y.K.; Chai, B.-H.; Chew, K.C.M.; Ang, E.-T.; Tsang, F.; Tan, B.W.Q.; Hong, E.T.E.; Asad, A.B.A.; Chuang, K.-H. Transgenic Mice Overexpressing the Divalent Metal Transporter 1 Exhibit Iron Accumulation and Enhanced Parkin Expression in the Brain. Neuromol. Med. 2017, 19, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.J.; Borquez, D.A.; Nunez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Díaz, S.; Estévez-Silva, H.; Orädd, G.; AfBjerken, S.; Marcellino, D.; Virel, A. An Altered Blood–Brain Barrier Contributes to Brain Iron Accumulation and Neuroinflammation in the 6-OHDA Rat Model of Parkinson’s Disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Marder, K.; Graziano, J.; Freyer, G.; Slavkovich, V.; LoIacono, N.; Cote, L.; Mayeux, R. Altered Systemic Iron Metabolism in Parkinson’s Disease. Neurology 1997, 49, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Ferese, R.; Scala, S.; Biagioni, F.; Giardina, E.; Zampatti, S.; Modugno, N.; Colonnese, C.; Storto, M.; Fornai, F.; Novelli, G. Heterozygous PLA2G6 Mutation Leads to Iron Accumulation within Basal Ganglia and Parkinson’s Disease. Front. Neurol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Torsdottir, G.; Kristinsson, J.; Sveinbjornsdottir, S.; Snaedal, J.; Johannesson, T. Copper, Ceruloplasmin, Superoxide Dismutase and Iron Parameters in Parkinson’s Disease. Pharmacol. Toxicol. 1999, 85, 239–243. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the Levels of Iron, Ferritin and Other Trace Metals in Parkinson’s Disease and Other Neurodegenerative Diseases Affecting the Basal Ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Pall, H.S.; Blake, D.R.; Gutteridge, J.M.; Williams, A.C.; Lunec, J.; Hall, M.; Taylor, A. Raised Cerebrospinal-Fluid Copper Concentration in Parkinson’s Disease. Lancet 1987, 330, 238–241. [Google Scholar] [CrossRef]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A. Patterns of Levels of Biological Metals in CSF Differ among Neurodegenerative Diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Arnal, N.; Cristalli, D.O.; de Alaniz, M.J.T.; Marra, C.A. Clinical Utility of Copper, Ceruloplasmin, and Metallothionein Plasma Determinations in Human Neurodegenerative Patients and Their First-Degree Relatives. Brain Res. 2010, 1319, 118–130. [Google Scholar] [CrossRef]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.M.; Reyes, S.; Halliday, G.M.; Mercer, J.F.B. Copper Pathology in Vulnerable Brain Regions in Parkinson’s Disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef] [Green Version]
- Montes, S.; Rivera-Mancia, S.; Diaz-Ruiz, A.; Tristan-Lopez, L.; Rios, C. Copper and Copper Proteins in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2014, 2014, 7251. [Google Scholar] [CrossRef] [Green Version]
- Howells, C.; West, A.K.; Chung, R.S. Neuronal Growth-inhibitory Factor (Metallothionein-3): Evaluation of the Biological Function of Growth-inhibitory Factor in the Injured and Neurodegenerative Brain. FEBS J. 2010, 277, 2931–2939. [Google Scholar] [CrossRef]
- Schober, A. Classic Toxin-Induced Animal Models of Parkinson’s Disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef]
- Bove, J.; Perier, C. Neurotoxin-Based Models of Parkinson’s Disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef]
- Cheng, L.; Quek, C.Y.J.; Hung, L.W.; Sharples, R.A.; Sherratt, N.A.; Barnham, K.J.; Hill, A.F. Gene Dysregulation Is Restored in the Parkinson’s Disease MPTP Neurotoxic Mice Model upon Treatment of the Therapeutic Drug Cu II (Atsm). Sci. Rep. 2016, 6, 22398. [Google Scholar]
- Gutbier, S.; Kyriakou, S.; Schildknecht, S.; Ückert, A.-K.; Brüll, M.; Lewis, F.; Dickens, D.; Pearson, L.; Elson, J.L.; Michel, S. Design and Evaluation of Bi-Functional Iron Chelators for Protection of Dopaminergic Neurons from Toxicants. Arch. Toxicol. 2020, 94, 3105–3123. [Google Scholar] [CrossRef]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.H.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. Cu II (Atsm) Improves the Neurological Phenotype and Survival of SOD1 G93A Mice and Selectively Increases Enzymatically Active SOD1 in the Spinal Cord. Sci. Rep. 2017, 7, 42292. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.; Fujibayashi, Y.; Tajmia, N.; Yokoyama, A. Cu-ATSM, an Intracellular-Accessible Superoxide Dismutase (SOD)-like Copper Complex: Evaluation in an Ischemia-Reperfusion Injury Model. Biol. Pharm. Bull. 1994, 17, 701–704. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Raposinho, P.; Campello, M.P.C.; Belo, D.; Guerreiro, J.F.; Alves, V.; Fonseca, A.; Abrunhosa, A.J.; Paulo, A.; Mendes, F. Anticancer Activity and Mode of Action of Copper (II)-Bis (Thiosemicarbazonato) Complexes with Pendant Nitrogen Heterocycles. Eur. J. Inorg. Chem. 2021, 91, 1337–1348. [Google Scholar] [CrossRef]
- Dearling, J.L.J.; Packard, A.B. Some Thoughts on the Mechanism of Cellular Trapping of Cu (II)-ATSM. Nucl. Med. Biol. 2010, 37, 237–243. [Google Scholar] [CrossRef]
- Fujibayashi, Y.; Taniuchi, H.; Yonekura, Y.; Ohtani, H. Copper-62-ATSM: A New Hypoxia Imaging Agent with High Membrane Permeability and Low Redox Potential. J. Nucl. Med. 1997, 38, 1155. [Google Scholar] [PubMed]
- Dehdashti, F.; Grigsby, P.W.; Mintun, M.A.; Lewis, J.S.; Siegel, B.A.; Welch, M.J. Assessing Tumor Hypoxia in Cervical Cancer by Positron Emission Tomography with 60Cu-ATSM: Relationship to Therapeutic Response—A Preliminary Report. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 1233–1238. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased Oxidative Stress Is Related to Disease Severity in the ALS Motor Cortex: A PET Study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S. The Hypoxia Imaging Agent CuII (Atsm) Is Neuroprotective and Improves Motor and Cognitive Functions in Multiple Animal Models of Parkinson’s Disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef] [PubMed]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saeidi, M.; Goudarzi, M.; Pourkarim, H.; Azad, M. A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239. [Google Scholar] [PubMed]
- Pinto, V.M.; Forni, G.L. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. Int. J. Mol. Sci. 2020, 21, 8771. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glickstein, H.; ben El, R.; Shvartsman, M.; Cabantchik, Z.I. Intracellular Labile Iron Pools as Direct Targets of Iron Chelators: A Fluorescence Study of Chelator Action in Living Cells. Blood 2005, 106, 3242–3250. [Google Scholar] [CrossRef]
- Sohn, Y.; Mitterstiller, A.; Breuer, W.; Weiss, G.; Cabantchik, Z.I. Rescuing Iron-overloaded Macrophages by Conservative Relocation of the Accumulated Metal. Br. J. Pharmacol. 2011, 164, 406–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Anwer, M.K.; Makeen, H.A.; Albratty, M.; Mohan, S.; Bungau, S. Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 678. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020678
Behl T, Madaan P, Sehgal A, Singh S, Anwer MK, Makeen HA, Albratty M, Mohan S, Bungau S. Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(2):678. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020678
Chicago/Turabian StyleBehl, Tapan, Piyush Madaan, Aayush Sehgal, Sukhbir Singh, Md Khalid Anwer, Hafiz A. Makeen, Mohammed Albratty, Syam Mohan, and Simona Bungau. 2022. "Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 2: 678. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020678