
Proteomic Profiling of Extracellular Vesicles Released by Leptin-Treated Breast Cancer Cells: A Potential Role in Cancer Metabolism
 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
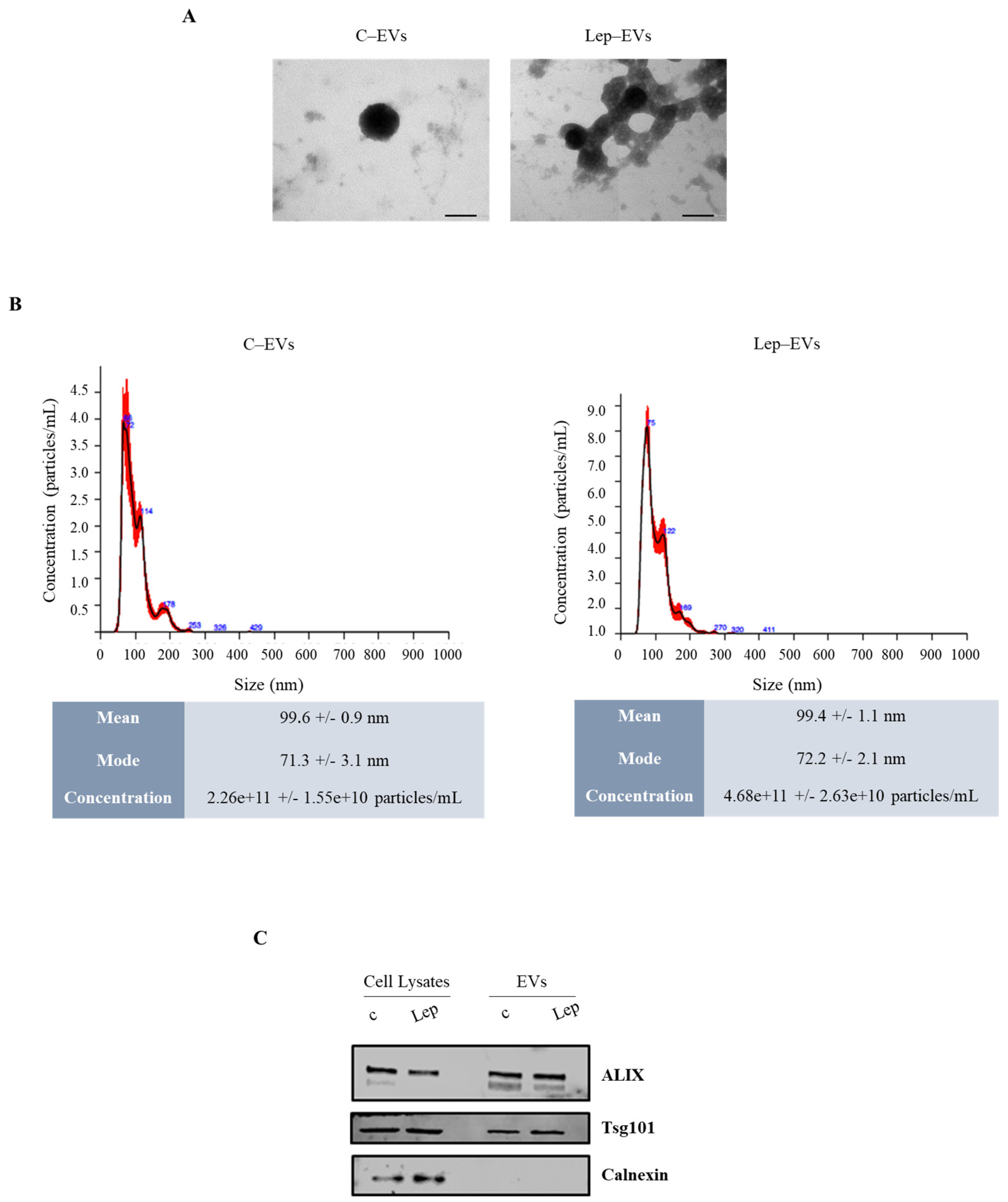
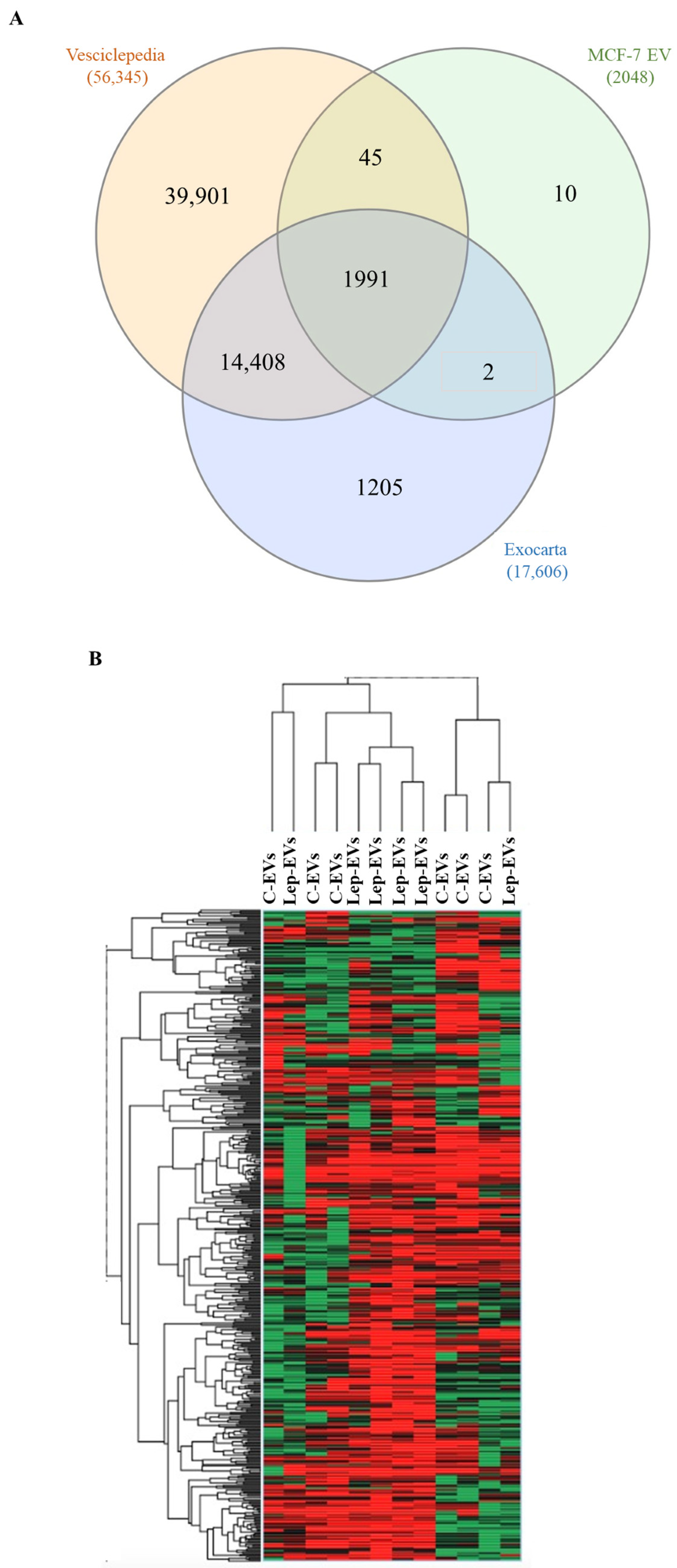
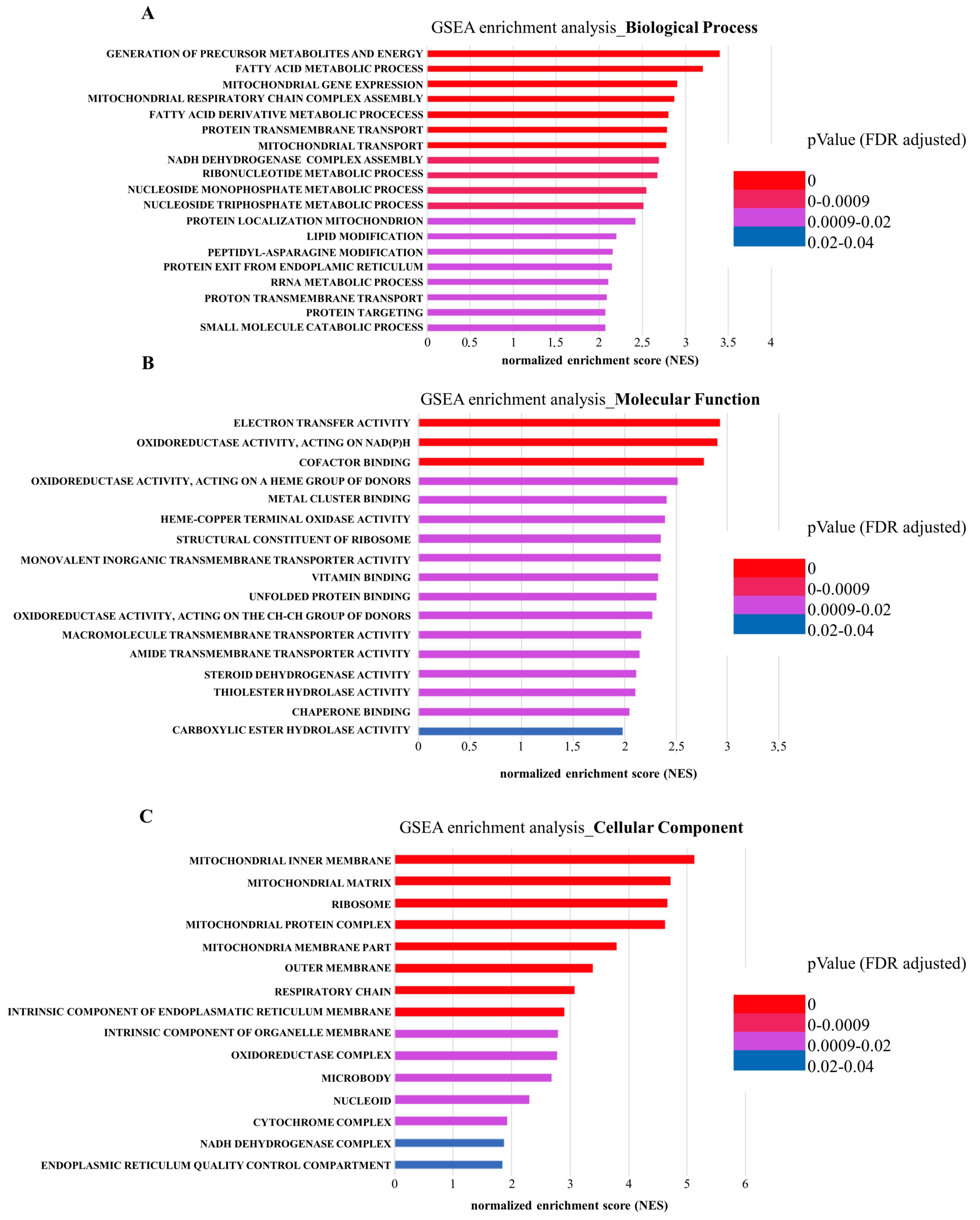
2.1. Extracellular Vesicles from Leptin-Treated MCF-7 Breast Cancer Cells Show an Enrichment in Proteins Involved in Energetic Metabolism
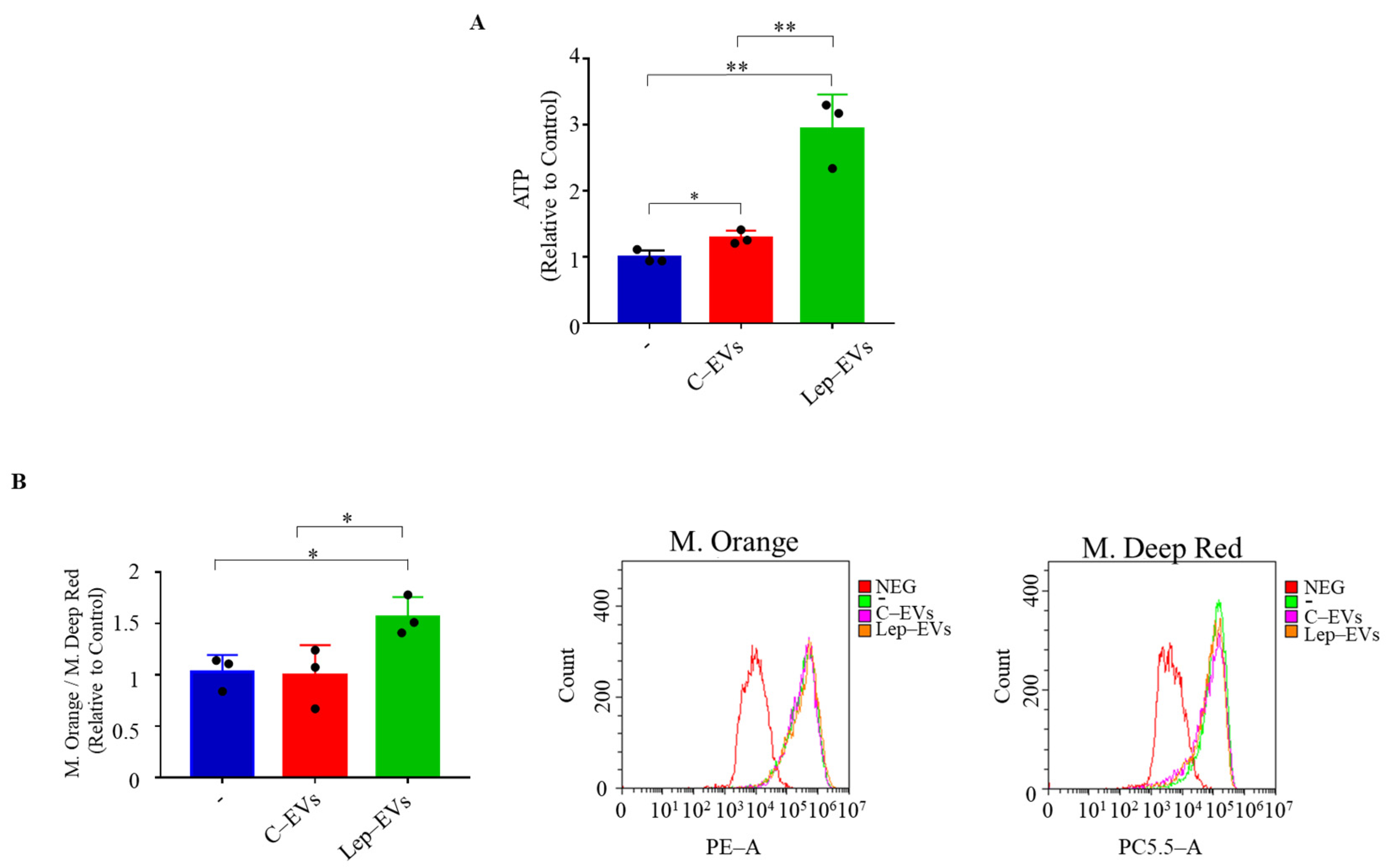
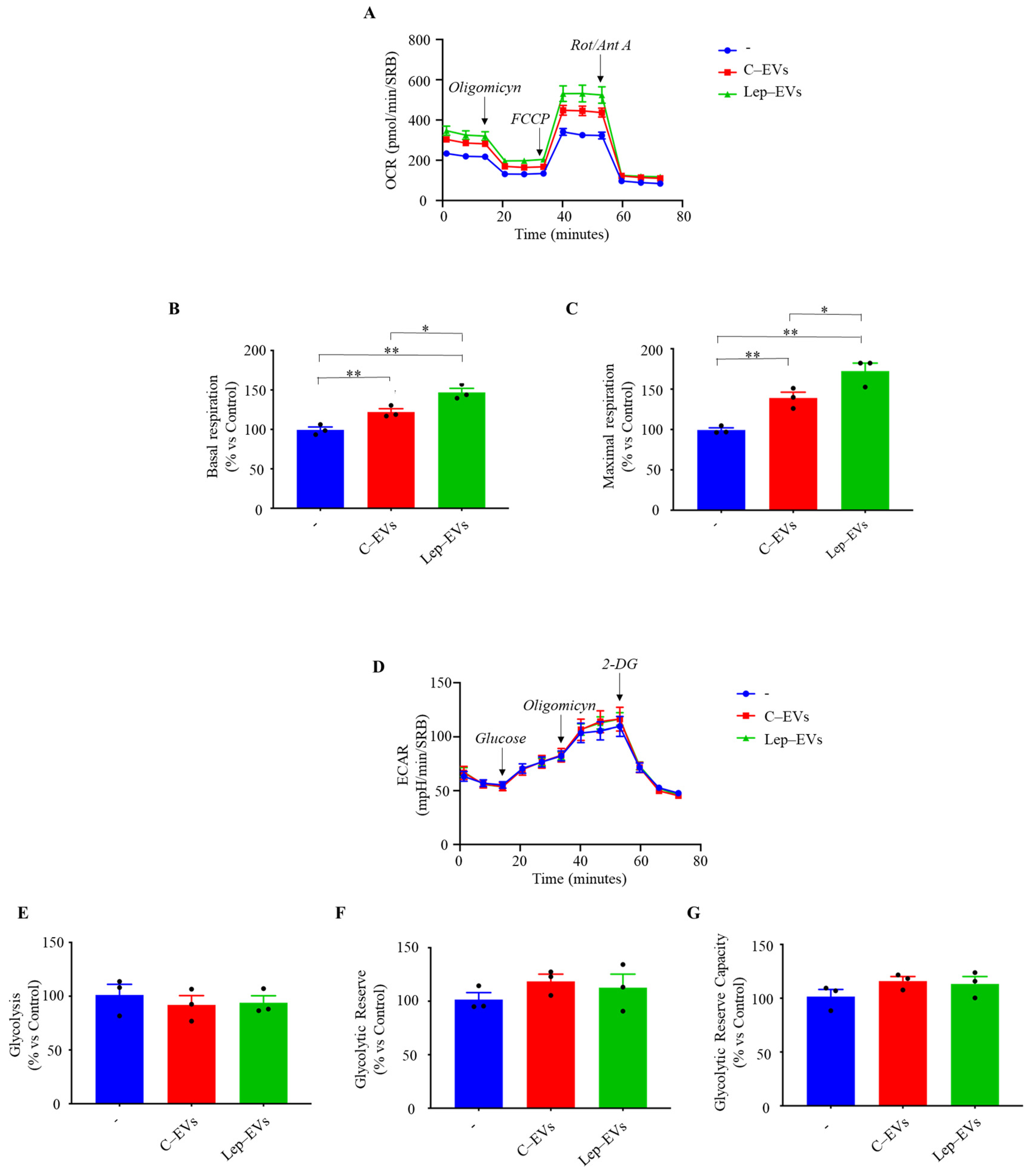
2.2. Extracellular Vesicles Released by Leptin-Treated MCF-7 Cells Sustain Mitochondrial Metabolism in Breast Cancer Cells
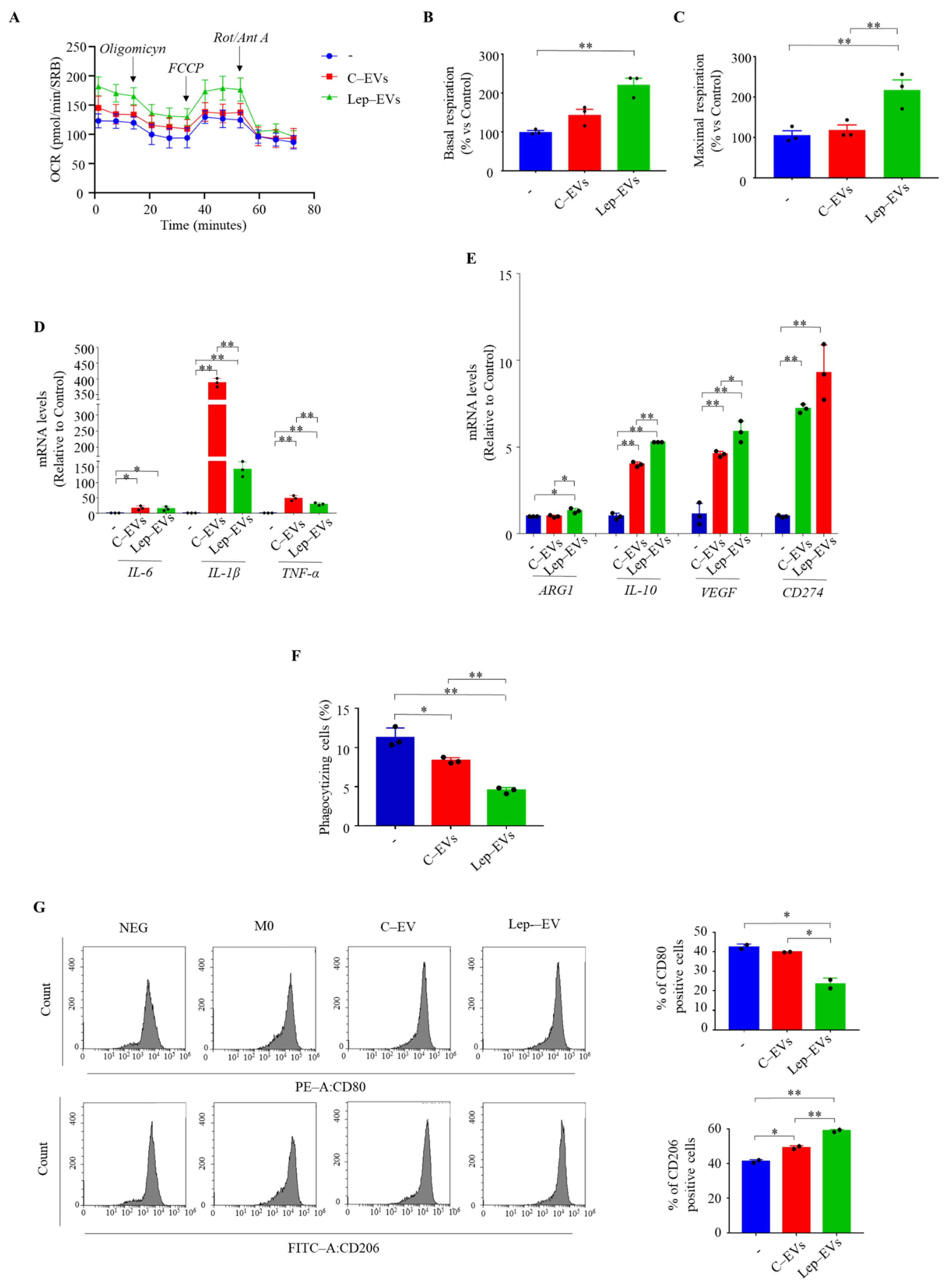
2.3. Extracellular Vesicles from Leptin-Treated MCF-7 Cells Influence Metabolic Profile of Macrophages
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Cultures
4.3. Isolation of Extracellular Vesicles
4.4. Transmission Electron Microscopy (TEM)
4.5. Nanoparticle Tracking Analysis (NTA)
4.6. Immunoblot Analysis
4.7. Proteomic Analysis
4.7.1. Protein Digestion
4.7.2. Protein Purification
4.7.3. Mass Spectometry Analysis
4.7.4. Data Analysis
4.8. Analysis of Extracellular Vesicles Proteome Cargoes
4.9. Functional Enrichment and Biological Network Analysis
4.10. Quantitation of Cellular ATP Levels
4.11. Evaluation of Mitochondrial Mass and Mitochondrial Membrane Potential
4.12. Seahorse XFe96 Metabolic Profile Analysis
4.13. Sulforhodamine B (SRB) Assay
4.14. Total RNA Extraction and Reverse Transcription Real-Time PCR Assay
4.15. Phagocytosis Assay
4.16. Flow Cytometry
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barone, I.; Giordano, C.; Bonofiglio, D.; Ando, S.; Catalano, S. The weight of obesity in breast cancer progression and metastasis: Clinical and molecular perspectives. Semin. Cancer Biol. 2020, 60, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Caruso, A.; Gelsomino, L.; Giordano, C.; Bonofiglio, D.; Catalano, S.; Ando, S. Obesity and endocrine therapy resistance in breast cancer: Mechanistic insights and perspectives. Obes. Rev. 2022, 23, e13358. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 4325–4331. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2012, 8, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Gramignano, G.; Floris, C.; Murenu, G.; Sollai, G.; Maccio, A. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J. Cell. Mol. Med. 2014, 18, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Maccio, A.; Madeddu, C.; Gramignano, G.; Mulas, C.; Floris, C.; Massa, D.; Astara, G.; Chessa, P.; Mantovani, G. Correlation of body mass index and leptin with tumor size and stage of disease in hormone-dependent postmenopausal breast cancer: Preliminary results and therapeutic implications. J. Mol. Med. 2010, 88, 677–686. [Google Scholar] [CrossRef]
- Hosney, M.; Sabet, S.; El-Shinawi, M.; Gaafar, K.M.; Mohamed, M.M. Leptin is overexpressed in the tumor microenvironment of obese patients with estrogen receptor positive breast cancer. Exp. Ther. Med. 2017, 13, 2235–2246. [Google Scholar] [CrossRef] [Green Version]
- Gelsomino, L.; Naimo, G.D.; Malivindi, R.; Augimeri, G.; Panza, S.; Giordano, C.; Barone, I.; Bonofiglio, D.; Mauro, L.; Catalano, S.; et al. Knockdown of Leptin Receptor Affects Macrophage Phenotype in the Tumor Microenvironment Inhibiting Breast Cancer Growth and Progression. Cancers 2020, 12, 2078. [Google Scholar] [CrossRef]
- Gelsomino, L.; Giordano, C.; Camera, G.; Sisci, D.; Marsico, S.; Campana, A.; Tarallo, R.; Rinaldi, A.; Fuqua, S.; Leggio, A.; et al. Leptin Signaling Contributes to Aromatase Inhibitor Resistant Breast Cancer Cell Growth and Activation of Macrophages. Biomolecules 2020, 10, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, C.; Gelsomino, L.; Barone, I.; Panza, S.; Augimeri, G.; Bonofiglio, D.; Rovito, D.; Naimo, G.D.; Leggio, A.; Catalano, S.; et al. Leptin Modulates Exosome Biogenesis in Breast Cancer Cells: An Additional Mechanism in Cell-to-Cell Communication. J. Clin. Med. 2019, 8, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, C.; La Camera, G.; Gelsomino, L.; Barone, I.; Bonofiglio, D.; Ando, S.; Catalano, S. The Biology of Exosomes in Breast Cancer Progression: Dissemination, Immune Evasion and Metastatic Colonization. Cancers 2020, 12, 2179. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther. Nucleic Acids 2020, 19, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Hernandez, O.; Serna-Marquez, N.; Castillo-Sanchez, R.; Salazar, E.P. Extracellular vesicles from MDA-MB-231 breast cancer cells stimulated with linoleic acid promote an EMT-like process in MCF10A cells. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 299–310. [Google Scholar] [CrossRef]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [Green Version]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-beta in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- La Camera, G.; Gelsomino, L.; Caruso, A.; Panza, S.; Barone, I.; Bonofiglio, D.; Ando, S.; Giordano, C.; Catalano, S. The Emerging Role of Extracellular Vesicles in Endocrine Resistant Breast Cancer. Cancers 2021, 13, 1160. [Google Scholar] [CrossRef]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and breast cancer drug resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef]
- Semina, S.E.; Scherbakov, A.M.; Kovalev, S.V.; Shevchenko, V.E.; Krasil’nikov, M.A. Horizontal Transfer of Tamoxifen Resistance in MCF-7 Cell Derivates: Proteome Study. Cancer Investig. 2017, 35, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Ciravolo, V.; Huber, V.; Ghedini, G.C.; Venturelli, E.; Bianchi, F.; Campiglio, M.; Morelli, D.; Villa, A.; Della Mina, P.; Menard, S.; et al. Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy. J. Cell. Physiol. 2012, 227, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Feng, T.; Zhang, P.; Sun, Y.; Wang, Y.; Tong, J.; Dai, H.; Hua, Z. High throughput sequencing identifies breast cancer-secreted exosomal LncRNAs initiating pulmonary pre-metastatic niche formation. Gene 2019, 710, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef] [PubMed]
- Rontogianni, S.; Synadaki, E.; Li, B.; Liefaard, M.C.; Lips, E.H.; Wesseling, J.; Wu, W.; Altelaar, M. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Commun. Biol. 2019, 2, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell. Biol. 2006, 3, 22. [Google Scholar] [CrossRef]
- Tiruvayipati, S.; Wolfgeher, D.; Yue, M.; Duan, F.F.; Andrade, J.; Jiang, H.; Schuger, L. Variability in protein cargo detection in technical and biological replicates of exosome-enriched extracellular vesicles. PLoS ONE 2020, 15, e0228871. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPARgamma Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174. [Google Scholar] [CrossRef]
- Liu, P.S.; Ho, P.C. Determining Macrophage Polarization upon Metabolic Perturbation. Methods Mol. Biol. 2019, 1862, 173–186. [Google Scholar] [CrossRef]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Pisanti, S.; Napolitano, F.; Di Somma, S.; Martinelli, R.; Portella, G. Tumor-Associated Macrophage Status in Cancer Treatment. Cancers 2020, 12, 1987. [Google Scholar] [CrossRef]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 242. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Pham, D.V.; Park, P.H. Tumor Metabolic Reprogramming by Adipokines as a Critical Driver of Obesity-Associated Cancer Progression. Int. J. Mol. Sci. 2021, 22, 1444. [Google Scholar] [CrossRef]
- Park, J.; Kusminski, C.M.; Chua, S.C.; Scherer, P.E. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am. J. Pathol. 2010, 177, 3133–3144. [Google Scholar] [CrossRef]
- Blanquer-Rossello, M.M.; Santandreu, F.M.; Oliver, J.; Roca, P.; Valle, A. Leptin Modulates Mitochondrial Function, Dynamics and Biogenesis in MCF-7 Cells. J. Cell. Biochem. 2015, 116, 2039–2048. [Google Scholar] [CrossRef]
- Blanquer-Rossello, M.D.M.; Oliver, J.; Sastre-Serra, J.; Valle, A.; Roca, P. Leptin regulates energy metabolism in MCF-7 breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 72, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sun, Y.; Fei, Z.; Yang, Z.; Duan, K.; Zi, J.; Cui, Q.; Yu, M.; Xiong, W. Leptin promotes fatty acid oxidation and OXPHOS via the c-Myc/PGC-1 pathway in cancer cells. Acta Biochim. Et Biophys. Sin. 2019, 51, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Amari, L.; Germain, M. Mitochondrial Extracellular Vesicles—Origins and Roles. Front. Mol. Neurosci. 2021, 14, 767219. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.V.; Tan, A.S. Effects of mitochondrial gene deletion on tumorigenicity of metastatic melanoma: Reassessing the Warburg effect. Rejuvenation Res. 2010, 13, 139–141. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Gao, Y.; Liu, J.; Huang, Y.; Yin, J.; Feng, Y.; Shi, L.; Meloni, B.P.; Zhang, C.; Zheng, M.; et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct. Target. Ther. 2021, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Ozsvari, B.; Sotgia, F.; Lisanti, M.P. High ATP Production Fuels Cancer Drug Resistance and Metastasis: Implications for Mitochondrial ATP Depletion Therapy. Front. Oncol. 2021, 11, 740720. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Kadel, S.; Townsend, M.G.; Oliwa, M.; Guerriero, J.L. Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front. Immunol. 2021, 12, 643771. [Google Scholar] [CrossRef] [PubMed]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Augimeri, G.; La Camera, G.; Gelsomino, L.; Giordano, C.; Panza, S.; Sisci, D.; Morelli, C.; Gyorffy, B.; Bonofiglio, D.; Ando, S.; et al. Evidence for Enhanced Exosome Production in Aromatase Inhibitor-Resistant Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5841. [Google Scholar] [CrossRef]
- De Amicis, F.; Giordano, F.; Vivacqua, A.; Pellegrino, M.; Panno, M.L.; Tramontano, D.; Fuqua, S.A.; Ando, S. Resveratrol, through NF-Y/p53/Sin3/HDAC1 complex phosphorylation, inhibits estrogen receptor alpha gene expression via p38MAPK/CK2 signaling in human breast cancer cells. FASEB J. 2011, 25, 3695–3707. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Prestagiacomo, L.E.; Gabriele, C.; Morelli, P.; Rota, M.A.; Alba, S.; Cuda, G.; Damiano, R.; Gaspari, M. Proteomic Profile of EPS-Urine through FASP Digestion and Data-Independent Analysis. J. Vis. Exp. 2021, 171, e62512. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Nigro, A.; Frattaruolo, L.; Fava, M.; De Napoli, I.; Greco, M.; Comande, A.; De Santo, M.; Pellegrino, M.; Ricci, E.; Giordano, F.; et al. Bortezomib-Loaded Mesoporous Silica Nanoparticles Selectively Alter Metabolism and Induce Death in Multiple Myeloma Cells. Cancers 2020, 12, 2709. [Google Scholar] [CrossRef] [PubMed]
- Panza, S.; Malivindi, R.; Chemi, F.; Rago, V.; Giordano, C.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Giordano, F.; Ando, S.; et al. Glucocorticoid Receptor as a Potential Target to Decrease Aromatase Expression and Inhibit Leydig Tumor Growth. Am. J. Pathol. 2016, 186, 1328–1339. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelsomino, L.; Barone, I.; Caruso, A.; Giordano, F.; Brindisi, M.; Morello, G.; Accattatis, F.M.; Panza, S.; Cappello, A.R.; Bonofiglio, D.; et al. Proteomic Profiling of Extracellular Vesicles Released by Leptin-Treated Breast Cancer Cells: A Potential Role in Cancer Metabolism. Int. J. Mol. Sci. 2022, 23, 12941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232112941
Gelsomino L, Barone I, Caruso A, Giordano F, Brindisi M, Morello G, Accattatis FM, Panza S, Cappello AR, Bonofiglio D, et al. Proteomic Profiling of Extracellular Vesicles Released by Leptin-Treated Breast Cancer Cells: A Potential Role in Cancer Metabolism. International Journal of Molecular Sciences. 2022; 23(21):12941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232112941
Chicago/Turabian StyleGelsomino, Luca, Ines Barone, Amanda Caruso, Francesca Giordano, Matteo Brindisi, Giovanna Morello, Felice Maria Accattatis, Salvatore Panza, Anna Rita Cappello, Daniela Bonofiglio, and et al. 2022. "Proteomic Profiling of Extracellular Vesicles Released by Leptin-Treated Breast Cancer Cells: A Potential Role in Cancer Metabolism" International Journal of Molecular Sciences 23, no. 21: 12941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232112941