
Identification of Active Compounds against Melanoma Growth by Virtual Screening for Non-Classical Human DHFR Inhibitors

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Generation and Validation of Pharmacophore Models and Preliminary Screening
2.2. Ensemble Generation and Docking-Based Screening
2.3. Clustering and Selection of Hit Compounds C1 and C2
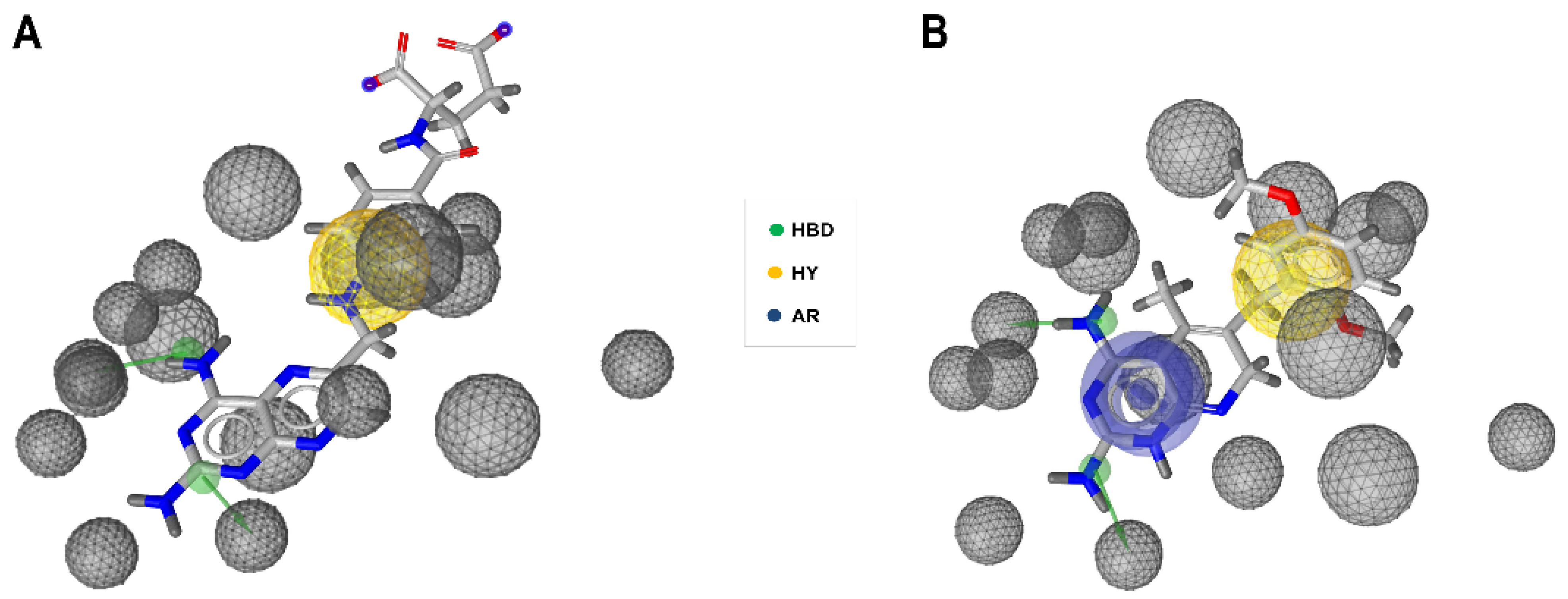
2.4. Re-Docking and Analysis of Binding Mode and Energy of C1 and C2
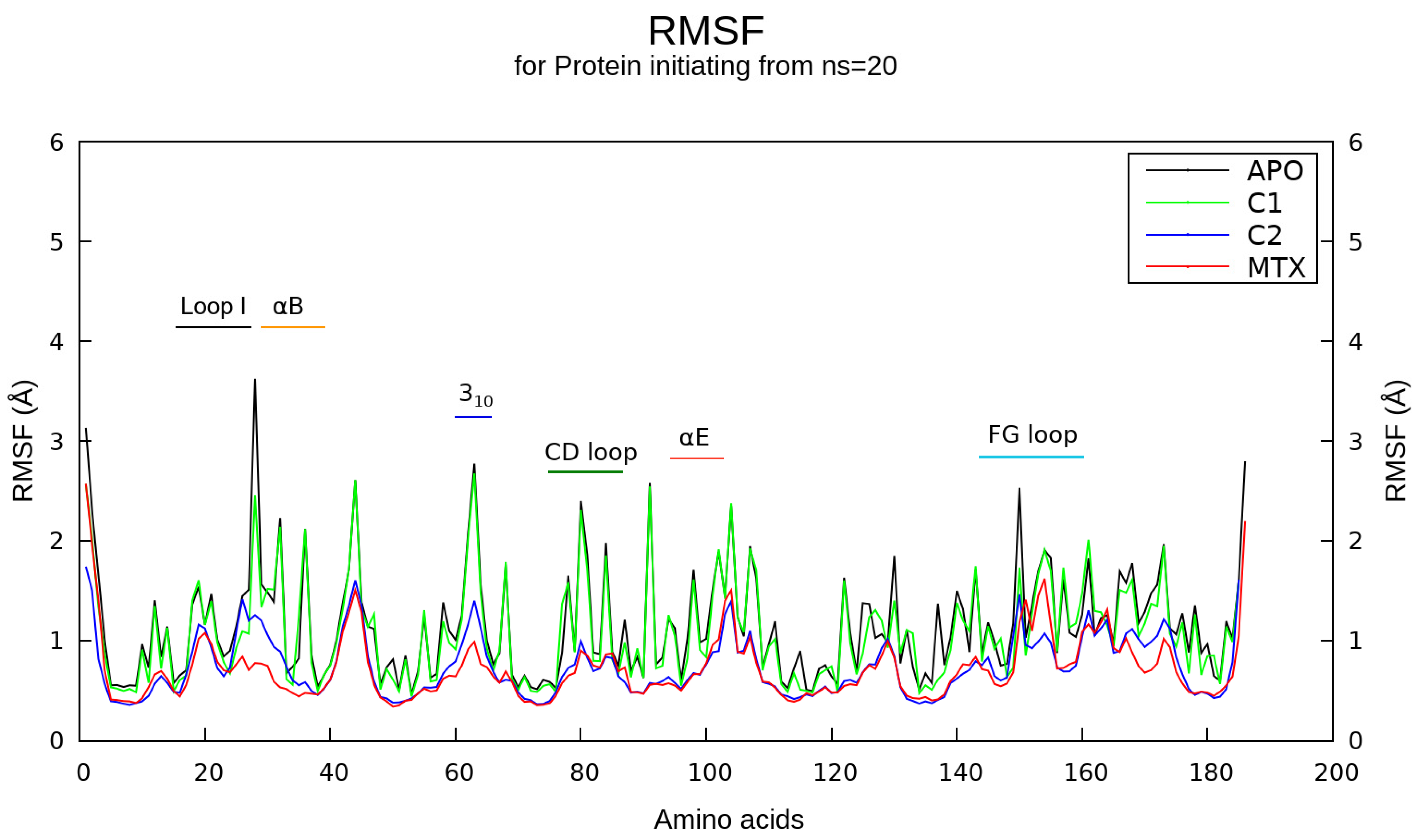
2.5. Molecular Dynamics Simulations
2.6. Binding Free Energy and DFT Analyses
2.7. ADME Profiling
2.8. Biological Assay on Melanoma Cells
3. Discussion
4. Materials and Methods
4.1. Pharmacophore Models Generation
4.2. Validation of Pharmacophore Hypotheses
4.3. Pharmacophore-Based Virtual Screening
4.4. Generation of Conformational Ensemble
4.5. Ensemble-Based Molecular Docking
4.6. Hit Dataset Clustering and Analysis
4.7. Re-Docking with Conserved Water Molecules and Binding Pose Comparison
4.8. Molecular Dynamics (MD) Simulations
4.9. Binding Free Energy Calculations
4.10. Density Functional Theory
4.11. Chemoinformatics Analysis
4.12. Cell Cultures and Experimental Design
4.13. Determination of Cell Viability by MTT and Trypan Blue Assays
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhagat, K.; Kumar, N.; Kaur Gulati, H.; Sharma, A.; Kaur, A.; Singh, J.V.; Singh, H.; Bedi, P.M.S. Dihydrofolate Reductase Inhibitors: Patent Landscape and Phases of Clinical Development (2001–2021). Expert Opin. Ther. Pat. 2022, 32, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Q.; Bennett, B.C.; Wymore, T.; Li, Z.; Wilson, M.A.; Brooks, C.L.; Langan, P.; Kovalevsky, A.; Dealwis, C.G. Capturing the Catalytic Proton of Dihydrofolate Reductase: Implications for General Acid—Base Catalysis. ACS Catal. 2021, 11, 5873–5884. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, L.M.; Dyson, H.J.; Wright, P.E. Side Chain Conformational Averaging in Human Dihydrofolate Reductase. Biochemistry 2014, 53, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Ko, A. Folate Antagonists. In Cancer Management in Man: Chemotheraphy, Biological Therapy, Hyperthermia and Supporting Measures (Cancer Growth and Progression); Minev, B.R., Ed.; Springer Science+Business Media B.V: New York, NY, USA, 2011; pp. 125–143. ISBN 9789048197040. [Google Scholar]
- Laanpere, M.; Altmäe, S.; Stavreus-Evers, A.; Nilsson, T.K.; Yngve, A.; Salumets, A. Folate-Mediated One-Carbon Metabolism and Its Effect on Female Fertility and Pregnancy Viability. Nutr. Rev. 2010, 68, 99–113. [Google Scholar] [CrossRef]
- Willson, J. Structural Study Could Aid Design of Antifolates. Nat. Rev. Cancer 2022, 22, 608. [Google Scholar] [CrossRef]
- Walling, J. From Methotrexate to Pemetrexed and beyond. A Review of the Pharmacodynamic and Clinical Properties of Antifolates. Invest. New Drugs 2006, 24, 37–77. [Google Scholar] [CrossRef]
- Hamed, K.M.; Dighriri, I.M.; Baomar, A.F.; Alharthy, B.T.; Alenazi, F.E.; Alali, G.H.; Alenazy, R.H.; Alhumaidi, N.T.; Alhulayfi, D.H.; Alotaibi, Y.B.; et al. Overview of Methotrexate Toxicity: A Comprehensive Literature Review. Cureus 2022, 14, e29518. [Google Scholar] [CrossRef]
- Uchihara, Y.; Komori, R.; Tago, K.; Tamura, H.; Funakoshi-Tago, M. Methotrexate Significantly Induces Apoptosis by Inhibiting STAT3 Activation in NPM-ALK-Positive ALCL Cells. Biochem. Pharmacol. 2019, 170, 113666. [Google Scholar] [CrossRef]
- Salem, I.M.; Mostafa, S.M.; Salama, I.; El-Sabbagh, O.I.; A H Hegazy, W.; Ibrahim, T.S. Human Dihydrofolate Reductase Inhibition Effect of 1-Phenylpyrazolo[3,4-d]Pyrimidines: Synthesis, Antitumor Evaluation and Molecular Modeling Study. Bioorg. Chem. 2022, 129, 106207. [Google Scholar] [CrossRef]
- Rodríguez-López, J.N.; Sanchez-del-Campo, L.; Saez-Ayala, M.; Montenegro, M.F.; Cabezas-Herrera, J. Novel Antifolates as Prodrugs for the Treatment of Melanoma. In Research on Melanoma—A Glimpse into Current Directions and Future Trends; Murph, M., Ed.; InTech: London, UK, 2011; pp. 101–124. ISBN 9789533072937. [Google Scholar]
- Sánchez-del-Campo, L.; Montenegro, M.F.; Cabezas-Herrera, J.; Rodríguez-López, J.N. The Critical Role of Alpha-Folate Receptor in the Resistance of Melanoma to Methotrexate. Pigment Cell Melanoma Res. 2009, 22, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tran, T.T.; Duong, K.T.; Nguyen, T.; Le, U.M. Options of Therapeutics and Novel Delivery Systems of Drugs for the Treatment of Melanoma. Mol. Pharm. 2022. [Google Scholar] [CrossRef] [PubMed]
- Matthews, N.H.; Wen-Qing, L.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W., Farma, J., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Kosary, C.L.; Altekruse, S.F.; Ruhl, J.; Lee, R.; Dickie, L. Clinical and Prognostic Factors for Melanoma of the Skin Using SEER Registries: Collaborative Stage Data Collection System, Version 1 and Version 2. Cancer 2014, 120, 3807–3814. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and Other Nonclassical Antifolates an Excellent Template for Searching Modifications of Dihydrofolate Reductase Enzyme Inhibitors. J. Antibiot. (Tokyo) 2020, 73, 5–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Subbagh, H.I.; Hassan, G.S.; El-Messery, S.M.; Al-Rashood, S.T.; Al-Omary, F.A.M.; Abulfadl, Y.S.; Shabayek, M.I. Nonclassical Antifolates, Part 5. Benzodiazepine Analogs as a New Class of DHFR Inhibitors: Synthesis, Antitumor Testing and Molecular Modeling Study. Eur. J. Med. Chem. 2014, 74, 234–245. [Google Scholar] [CrossRef]
- Dewar, S.; Sienkiewicz, N.; Ong, H.B.; Wall, R.J.; Horn, D.; Fairlamb, X.A.H. The Role of Folate Transport in Antifolate Drug Action in Trypanosoma Brucei. J. Biol. Chem. 2016, 291, 24768–24778. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, H.; Shen, C.; Hu, X.; Gao, J.; Li, D.; Cao, D.; Hou, T. Combined Strategies in Structure-Based Virtual Physical Chemistry Chemical Physics Accepted Manuscript. Phys. Chem. Chem. Phys. 2020, 1–30. [Google Scholar] [CrossRef]
- Slater, O.; Kontoyianni, M.; Slater, O.; Kontoyianni, M. The Compromise of Virtual Screening and Its Impact on Drug Discovery The Compromise of Virtual Screening and Its Impact on Drug Discovery. Expert Opin. Drug Discov. 2019, 14, 1–19. [Google Scholar] [CrossRef]
- da Silva Rocha, S.F.L.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M.R. Virtual Screening Techniques in Drug Discovery: Review and Recent Applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Biswas, R.; Chowdhury, N.; Mukherjee, R.; Bagchi, A. Identification and Analyses of Natural Compounds as Potential Inhibitors of TRAF6-Basigin Interactions in Melanoma Using Structure-Based Virtual Screening and Molecular Dynamics Simulations. J. Mol. Graph. Model. 2018, 85, 281–293. [Google Scholar] [CrossRef]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.P.A.; McGuire, R.; Roumen, L.; de Esch, I.J.P.; de Vlieg, J.; Klomp, J.P.G.; de Graaf, C. From the Protein’s Perspective: The Benefits and Challenges of Protein Structure-Based Pharmacophore Modeling. Med. Chem. Commun. 2012, 3, 28–38. [Google Scholar] [CrossRef]
- Wallach, I. Pharmacophore Inference and Its Application to Computational Drug Discovery. Drug Dev. Res. 2011, 72, 17–25. [Google Scholar] [CrossRef]
- Ballante, F.; Kooistra, A.J.; Kampen, S.; De Graaf, C.; Carlsson, J. Structure-Based Virtual Screening for Ligands of G Protein-Coupled Receptors: What Can Molecular Docking Do for You? Pharmacol. Rev. 2022, 73, 1698–1736. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Chen, W.; Xiao, D.; Wang, C. Computational Molecular Docking and Virtual Screening Revealed Promising SARS-CoV-2 Drugs. Precis. Clin. Med. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- Stanzione, F.; Giangreco, I.; Cole, J.C. Use of Molecular Docking Computational Tools in Drug Discovery. In Progress in Medicinal Chemistry; Elsevier B.V.: Amsterdam, The Netherlands, 2021; Volume 60, pp. 273–343. [Google Scholar]
- Hernández-Rodríguez, M.; Rosales-Hernández, M.C.; Mendieta-Wejebe, J.E.; Martínez-Archundia, M.; Basurto, J.C. Current Tools and Methods in Molecular Dynamics (MD) Simulations for Drug Design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef]
- Onyango, H.; Odhiambo, P.; Angwenyi, D.; Okoth, P. In Silico Identification of New Anti-SARS-CoV-2 Main Protease (M(pro)) Molecules with Pharmacokinetic Properties from Natural Sources Using Molecular Dynamics (MD) Simulations and Hierarchical Virtual Screening. J. Trop. Med. 2022, 2022, 3697498. [Google Scholar] [CrossRef]
- Al-Shar’i, N.; Musleh, S.S. CHK1 Kinase Inhibition: Identification of Allosteric Hits Using MD Simulations, Pharmacophore Modeling, Docking and MM-PBSA Calculations. Mol. Divers. 2022, 26, 903–921. [Google Scholar] [CrossRef]
- Starosyla, S.A.; Volynets, G.P.; Protopopov, M.V.; Bdzhola, V.G.; Pashevin, D.O.; Polishchuk, V.O.; Kozak, T.O.; Stroi, D.O.; Dosenko, V.E.; Yarmoluk, S.M. Pharmacophore Modeling, Docking and Molecular Dynamics Simulation for Identification of Novel Human Protein Kinase C Beta (PKCβ) Inhibitors. Struct. Chem. 2022, 1–15. [Google Scholar] [CrossRef]
- Thangavel, N.; Albratty, M. Pharmacophore Model-Aided Virtual Screening Combined with Comparative Molecular Docking and Molecular Dynamics for Identification of Marine Natural Products as SARS-CoV-2 Papain-like Protease Inhibitors. Arab. J. Chem. 2022, 15, 104334. [Google Scholar] [CrossRef]
- Dhameliya, T.M.; Nagar, P.R.; Gajjar, N.D. Systematic Virtual Screening in Search of SARS CoV-2 Inhibitors against Spike Glycoprotein: Pharmacophore Screening, Molecular Docking, ADMET Analysis and MD Simulations. Mol. Divers. 2022, 26, 2775–2792. [Google Scholar] [CrossRef] [PubMed]
- Klon, A.E.; Héroux, A.; Ross, L.J.; Pathak, V.; Johnson, C.A.; Piper, J.R.; Borhani, D.W. Atomic Structures of Human Dihydrofolate Reductase Complexed with NADPH and Two Lipophilic Antifolates at 1.09 Å and 1.05 Å Resolution. J. Mol. Biol. 2002, 320, 677–693. [Google Scholar] [CrossRef]
- Cody, V.; Luft, J.R.; Pangborn, W. Understanding the Role of Leu22 Variants in Methotrexate Resistance: Comparison of Wild-Type and Leu22Arg Variant Mouse and Human Dihydrofolate Reductase Ternary Crystal Complexes with Methotrexate and NADPH. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Cody, V.; Piraino, J.; Pace, J.; Li, W.; Gangjee, A. Preferential Selection of Isomer Binding from Chiral Mixtures: Alternate Binding Modes Observed for the E and Z Isomers of a Series of 5-Substituted 2,4-Diaminofuro[2,3-d]Pyrimidines as Ternary Complexes with NADPH and Human Dihydrofolate Reductase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1271–1277. [Google Scholar] [CrossRef]
- Penhallurick, R.W.; Durnal, M.D.; Harold, A.; Ichiye, T. Adaptations for Pressure and Temperature in Dihydrofolate Reductases. Microorganisms 2021, 9, 1706. [Google Scholar] [CrossRef]
- Rizzi, A.; Fioni, A. Virtual Screening Using PLS Discriminant Analysis and ROC Curve Approach: An Application Study on PDE4 Inhibitors. J. Chem. Inf. Model. 2008, 48, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Al-Nadaf, A.H.; Taha, M.O. Discovery of New Renin Inhibitory Leads via Sequential Pharmacophore Modeling, QSAR Analysis, in Silico Screening and in Vitro Evaluation. J. Mol. Graph. Model. 2011, 29, 843–864. [Google Scholar] [CrossRef]
- Kim, H.J.; Choo, H.; Cho, Y.S.; No, K.T.; Pae, A.N. Novel GSK-3β Inhibitors from Sequential Virtual Screening. Bioorg. Med. Chem. 2008, 16, 636–643. [Google Scholar] [CrossRef]
- Bowman, A.L.; Lerner, M.G.; Carlson, H.A. Protein Flexibility and Species Specificity in Structure-Based Drug Discovery: Dihydrofolate Reductase as a Test System. J. Am. Chem. Soc. 2007, 129, 3634–3640. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Kraut, J. Loop and Subdomain Movements in the Mechanism of Escherichia Coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef]
- Shrimpton, P.; Mullaney, A.; Allemann, R.K. Functional Role for Tyr 31 in the Catalytic Cycle of Chicken Dihydrofolate Reductase. Proteins Struct. Funct. Genet. 2003, 51, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Wright, D.L. The Design and Docking of Virtual Compound Libraries to Structures of Drug Targets. Curr. Comput.—Aided Drug Des. 2005, 1, 103–127. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.F. The Impact of Aromatic Ring Count on Compound Developability—Are Too Many Aromatic Rings a Liability in Drug Design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, X.; Kisliuk, R.L.; Piraino, J.; Cody, V.; Gangjee, A. Design, Synthesis, Biological Evaluation and X-Ray Crystal Structure of Novel Classical 6,5,6-Tricyclic Benzo[4,5]Thieno[2,3-d]Pyrimidines as Dual Thymidylate Synthase and Dihydrofolate Reductase Inhibitors. Bioorg. Med. Chem. 2011, 19, 3585–3594. [Google Scholar] [CrossRef]
- Göksel, Y.; Zor, K.; Rindzevicius, T.; Thorhauge Als-Nielsen, B.E.; Schmiegelow, K.; Boisen, A. Quantification of Methotrexate in Human Serum Using Surface-Enhanced Raman Scattering—Toward Therapeutic Drug Monitoring. ACS Sens. 2021, 6, 2664–2673. [Google Scholar] [CrossRef]
- Panecka-Hofman, J.; Pöhner, I.; Spyrakis, F.; Zeppelin, T.; Di Pisa, F.; Dello Iacono, L.; Bonucci, A.; Quotadamo, A.; Venturelli, A.; Mangani, S.; et al. Comparative Mapping of On-Targets and off-Targets for the Discovery of Anti-Trypanosomatid Folate Pathway Inhibitors. Biochim. Biophys. Acta—Gen. Subj. 2017, 1861, 3215–3230. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Peltason, L.; Bajorath, J. Quo Vadis, Virtual Screening? A Comprehensive Survey of Prospective Applications. J. Med. Chem. 2010, 53, 8461–8467. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical Investigations on the HOMO–LUMO Gap and Global Reactivity Descriptor Studies, Natural Bond Orbital, and Nucleus-Independent Chemical Shifts Analyses of 3-Phenylbenzo[d]Thiazole-2(3H)-Imine and Its Para-Substituted Derivatives: Solvent and Subs. J. Chem. Res. 2021, 45, 147–158. [Google Scholar] [CrossRef]
- Pegu, D.; Deb, J.; Van Alsenoy, C.; Sarkar, U. Theoretical Investigation of Electronic, Vibrational, and Nonlinear Optical Properties of 4-Fluoro-4-Hydroxybenzophenone. Spectrosc. Lett. 2017, 50, 232–243. [Google Scholar] [CrossRef]
- Wróbel, A.; Baradyn, M.; Ratkiewicz, A.; Drozdowska, D. Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-targeted Compounds: Dihydrofolate Reductase (Dhfr) Inhibitors and Dna-binding Agents. Int. J. Mol. Sci. 2021, 22, 3685. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Tata, R.B.; Bishop, Ö.T. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules 2020, 25, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanović, M.; Gruden-Pavlović, M.; Zlatović, M. Stabilizing Non-Covalent Interactions of Ligand Aromatic Moieties and Proline in Ligand-Protein Systems. Monatshefte fur Chemie 2015, 146, 389–397. [Google Scholar] [CrossRef]
- Zondlo, N.J. Aromatic-Proline Interactions: Electronically Tunable CH/π Interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [Green Version]
- Biedermannova, L.; E. Riley, K.; Berka, K.; Hobza, P.; Vondrasek, J. Another Role of Proline: Stabilization Interactions in Proteins and Protein Complexes Concerning Proline and Tryptophane. Phys. Chem. Chem. Phys. 2008, 10, 6350–6359. [Google Scholar] [CrossRef]
- Leitgeb, B.; Tóth, G. Aromatic-Aromatic and Proline-Aromatic Interactions in Endomorphin-1 and Endomorphin-2. Eur. J. Med. Chem. 2005, 40, 674–686. [Google Scholar] [CrossRef]
- Baghel, P.; Roy, A.; Verma, S.; Satapathy, T.; Bahadur, S. Amelioration of Lipophilic Compounds in Regards to Bioavailability as Self-Emulsifying Drug Delivery System (SEDDS). Futur. J. Pharm. Sci. 2020, 6, 21. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Effect of Lipophilicity on the Bioavailability of Drugs After Percutaneous Administration by Dissolving Microneedles. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Orzeszko, B.; Kazimierczuk, Z.; Maurin, J.K.; Laudy, A.E.; Starościak, B.J.; Vilpo, J.; Vilpo, L.; Balzarini, J.; Orzeszko, A. Novel Adamantylated Pyrimidines and Their Preliminary Biological Evaluations. Farm. (Società Chim. Ital. 1989) 2004, 59, 929–937. [Google Scholar] [CrossRef]
- Zahid, M.; Yasin, K.A.; Akhtar, T.; Rama, N.H.; Hameed, S.; Al-Masoudi, N.A.; Loddo, R.; La Colla, P. Synthesis and in Vitro Antiproliferative Activity of New Adamantylthiazolyl-1,3,4-Oxadiazoles. Ark. Online J. Org. Chem. 2009, 11, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Gangjee, A.; Jain, H.; Kurup, S. Recent Advances in Classical and Non-Classical Antifolates as Antitumor and Antiopportunistic Infection Agents: Part I. Anticancer Agents Med. Chem. 2007, 7, 524–542. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Jain, H.; Kurup, S. Recent Advances in Classical and Non-Classical Antifolates as Antitumor and Antiopportunistic Infection Agents: Part II. Anticancer Agents Med. Chem. 2008, 8, 205–231. [Google Scholar] [CrossRef] [PubMed]
- Scocchera, E.; Reeve, S.M.; Keshipeddy, S.; Lombardo, M.N.; Hajian, B.; Sochia, A.E.; Alverson, J.B.; Priestley, N.D.; Anderson, A.C.; Wright, D.L. Charged Nonclassical Antifolates with Activity Against Gram-Positive and Gram-Negative Pathogens. ACS Med. Chem. Lett. 2016, 7, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, J.D.; Chudasama, C.J.; Patel, K.D. Diarylpyrazole Ligated Dihydropyrimidine Hybrids as Potent Non-Classical Antifolates and Their Efficacy Against Plasmodium Falciparum. Arch. Pharm. (Weinheim) 2017, 350, 1700088. [Google Scholar] [CrossRef]
- Liu, H.; Qin, Y.; Zhai, D.; Zhang, Q.; Gu, J.; Tang, Y.; Yang, J.; Li, K.; Yang, L.; Chen, S.; et al. Antimalarial Drug Pyrimethamine Plays a Dual Role in Antitumor Proliferation and Metastasis through Targeting DHFR and TP. Mol. Cancer Ther. 2019, 18, 541–555. [Google Scholar] [CrossRef] [Green Version]
- McGuire, J.J. Anticancer Antifolates: Current Status and Future Directions. Curr. Pharm. Des. 2003, 9, 2593–2613. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.E.J.; Dean, P.M. Three-Dimensional Hydrogen-Bond Geometry and Probability Information from a Crystal Survey. J. Comput. Aided. Mol. Des. 1996, 10, 607–622. [Google Scholar] [CrossRef]
- Tsai, J.; Taylor, R.; Chothia, C.; Gerstein, M. The Packing Density in Proteins: Standard Radii and Volumes. J. Mol. Biol. 1999, 290, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [Green Version]
- Kokh, D.B.; Wade, R.C.; Wenzel, W. Receptor Flexibility in Small-Molecule Docking Calculations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 298–314. [Google Scholar] [CrossRef]
- Dunbrack, R.L., Jr. Rotamer Libraries in the 21st Century. Curr. Opin. Struct. Biol. 2002, 12, 431–440. [Google Scholar] [CrossRef]
- Scouras, A.D.; Daggett, V. The Dynameomics Rotamer Library: Amino Acid Side Chain Conformations and Dynamics from Comprehensive Molecular Dynamics Simulations in Water. Protein Sci. 2011, 20, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The Penultimate Rotamer Library. Proteins Struct. Funct. Bioinform. 2000, 40, 389–408. [Google Scholar] [CrossRef]
- Janin, J.; Wodak, S.; Levitt, M.; Maigret, B. Conformation of Amino Acid Side-Chains in Proteins. J. Mol. Biol. 1978, 125, 357–386. [Google Scholar] [CrossRef]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble Docking into Multiple Crystallographically Derived Protein Structures: An Evaluation Based on the Statistical Analysis of Enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. Four-Dimensional Docking: A Fast and Accurate Account of Discrete Receptor Flexibility in Ligand Docking. J. Med. Chem. 2009, 52, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Malinauskas, T. ADL High-Throughput Molecular Docking Using Free Tools ZINC 8, AutoDockTools 1.5.2 and Docker 1.0. Available online: https://web.archive.org/web/20090410151007/http://users.ox.ac.uk/~jesu1458/docker/ (accessed on 15 May 2013).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine Tools: An Online Service for Analyzing and Clustering Small Molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 December 2016).
- Willett, P. Similarity-Based Virtual Screening Using 2D Fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Sander, T. Openmolecules.Org. Available online: http://www.openmolecules.org/propertyexplorer/applet.html (accessed on 13 December 2016).
- Kumar, A.; Zhang, K.Y.J. Investigation on the Effect of Key Water Molecules on Docking Performance in CSARdock Exercise. J. Chem. Inf. Model. 2013, 53, 1880–1892. [Google Scholar] [CrossRef]
- Thilagavathi, R.; Mancera, R.L. Ligand-Protein Cross-Docking with Water Molecules. J. Chem. Inf. Model. 2010, 50, 415–421. [Google Scholar] [CrossRef]
- Hendlich, M.; Bergner, A.; Günther, J.; Klebe, G. Relibase: Design and Development of a Database for Comprehensive Analysis of Protein-Ligand Interactions. J. Mol. Biol. 2003, 326, 607–620. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Berk, H.; Erik, L. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; EASC 2014. Lecture Notes in Computer Science; Springer International Publishing: Basel, Switzerland, 2015; Volume 8759, pp. 3–27. ISBN 9783319159751. [Google Scholar]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Meza, J.C. Steepest Descent. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 719–722. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Fias, S.; Van Damme, S.; Bultinck, P. Multidimensionality of Delocalization Indices and Nucleus Independent Chemical Shifts in Polycyclic Aromatic Hydrocarbons. J. Comput. Chem. 2008, 29, 358–366. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Source, O.; Discovery, D.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolotti, L.J.; Flurchick, K. An Introduction to Density Functional Theory. Rev. Comput. Chem. 2007, 7, 187–216. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for Novel Inhibitors Showing Back-to-Front Approach against VEGFR-2 Kinase Domain (4AG8) Employing Molecular Docking Mechanism and Molecular Dynamics Simulations. BMC Cancer 2018, 18, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Fatima, S.; Gupta, P.; Sharma, S.; Sharma, A.; Agarwal, S.M. ADMET Profiling of Geographically Diverse Phytochemical Using Chemoinformatic Tools. Future Med. Chem. 2019, 12, 69–87. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2001, 44, 235–249. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Jasial, S.; Hu, Y.; Bajorath, J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Gomez, L.A.; Alekseev, A.E.; Aleksandrova, L.A.; Brady, P.A.; Terzic, A. Use of the MTT Assay in Adult Ventricular Cardiomyocytes to Assess Viability: Effects of Adenosine and Potassium on Cellular Survival. J. Mol. Cell. Cardiol. 1997, 29, 1255–1266. [Google Scholar] [CrossRef]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. In Cancer Cell Culture: Methods and Protocols; Methods in Molecular Biology; Cree, I.A., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2011; Volume 731, pp. 237–245. ISBN 978-1-61779-079-9. [Google Scholar]
- Louis, K.S.; Siegel, A.C. Cell Viability Analysis Using Trypan Blue: Manual and Automated Methods. In Mammalian Cell Viability; Methods in Molecular Biology; Stoddart, M.J., Ed.; Humana Press Inc.: New York, NY, USA, 2011; Volume 740, pp. 7–12. ISBN 978-1-61779-107-9. [Google Scholar]
- Dawson, C.W.; Young, L.S. In Vitro Assays to Study Epithelial Cell Growth. In Epstein-Barr Virus Protocols; Methods in Molecular Biology; Wilson, J.B., May, G.H.W., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2001; Volume 174, pp. 165–172. [Google Scholar]
- Dong, X.; Xinglu, Z.; Jing, H.; Chen, J.; Liu, T.; Yang, B.; He, Q.; Yongzhou, H. Pharmacophore Identification, Virtual Screening and Biological Evaluation of Prenylated Flavonoids Derivatives as PKB/Akt1 Inhibitors. Eur. J. Med. Chem. 2011, 46, 5949–5958. [Google Scholar] [CrossRef]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Ligand and Structure Based Pharmacophore Modeling to Facilitate Novel Histone Deacetylase 8 Inhibitor Design. Eur. J. Med. Chem. 2010, 45, 4409–4417. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Property | Hit Dataset a | C1 | C2 |
|---|---|---|---|
| MW (Da) | 3.766 | 351.43 | 452.47 |
| LogP | 3.544 | 4.260 | 4.800 |
| Number of rings | 3.421 | 3 | 6 |
| Number of aromatic rings | 2.537 | 3 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vásquez, A.F.; Gómez, L.A.; González Barrios, A.; Riaño-Pachón, D.M. Identification of Active Compounds against Melanoma Growth by Virtual Screening for Non-Classical Human DHFR Inhibitors. Int. J. Mol. Sci. 2022, 23, 13946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232213946
Vásquez AF, Gómez LA, González Barrios A, Riaño-Pachón DM. Identification of Active Compounds against Melanoma Growth by Virtual Screening for Non-Classical Human DHFR Inhibitors. International Journal of Molecular Sciences. 2022; 23(22):13946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232213946
Chicago/Turabian StyleVásquez, Andrés Felipe, Luis Alberto Gómez, Andrés González Barrios, and Diego M. Riaño-Pachón. 2022. "Identification of Active Compounds against Melanoma Growth by Virtual Screening for Non-Classical Human DHFR Inhibitors" International Journal of Molecular Sciences 23, no. 22: 13946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232213946