
Imaging Methods Applicable in the Diagnostics of Alzheimer’s Disease, Considering the Involvement of Insulin Resistance
 , ,
, ,
Abstract
:1. Alzheimer’s Disease
2. Neuroimaging in the Cognitive Impairment
2.1. Radiological Neuroimaging
2.1.1. CT Neuroimaging
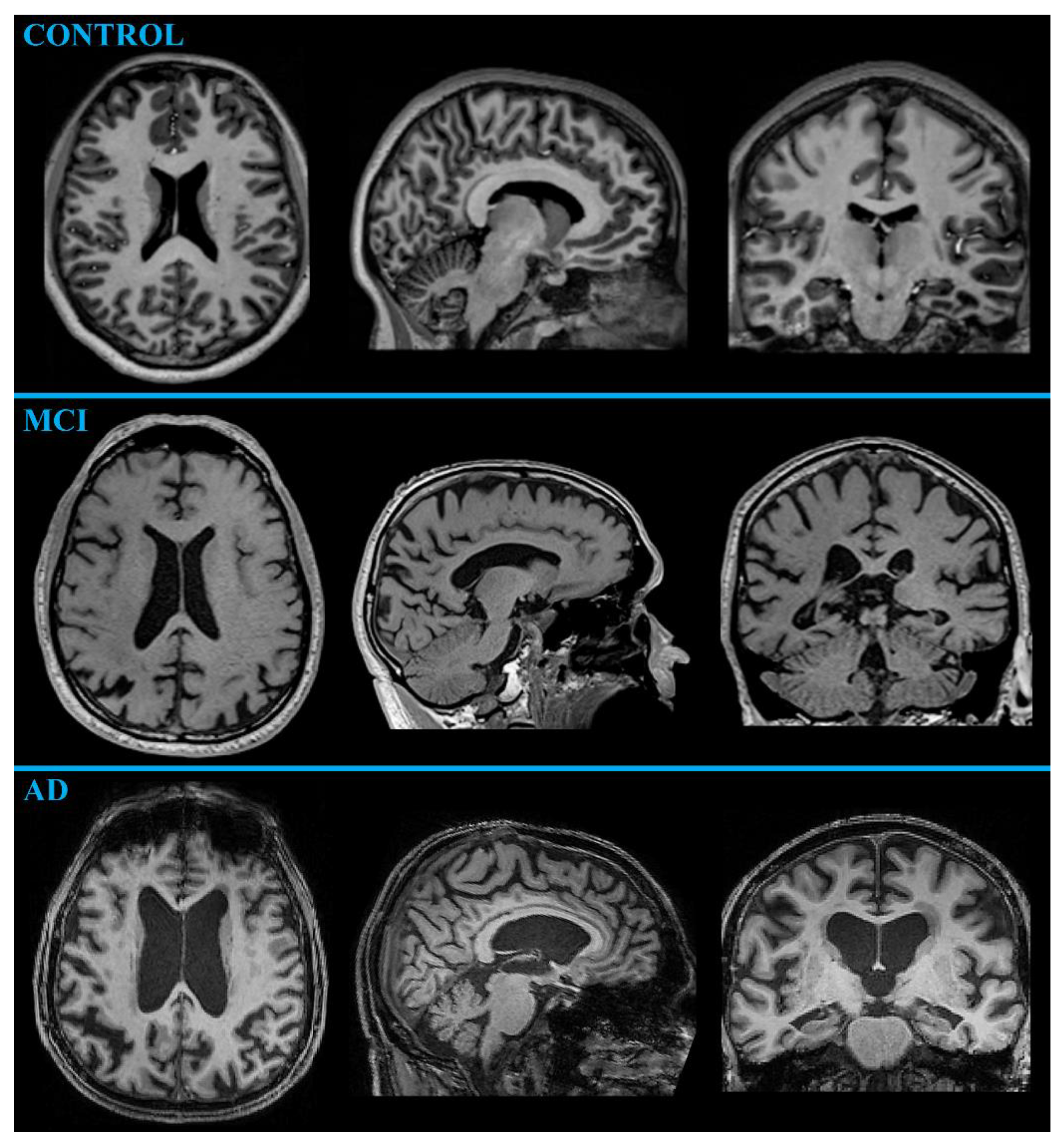
2.1.2. MR Neuroimaging
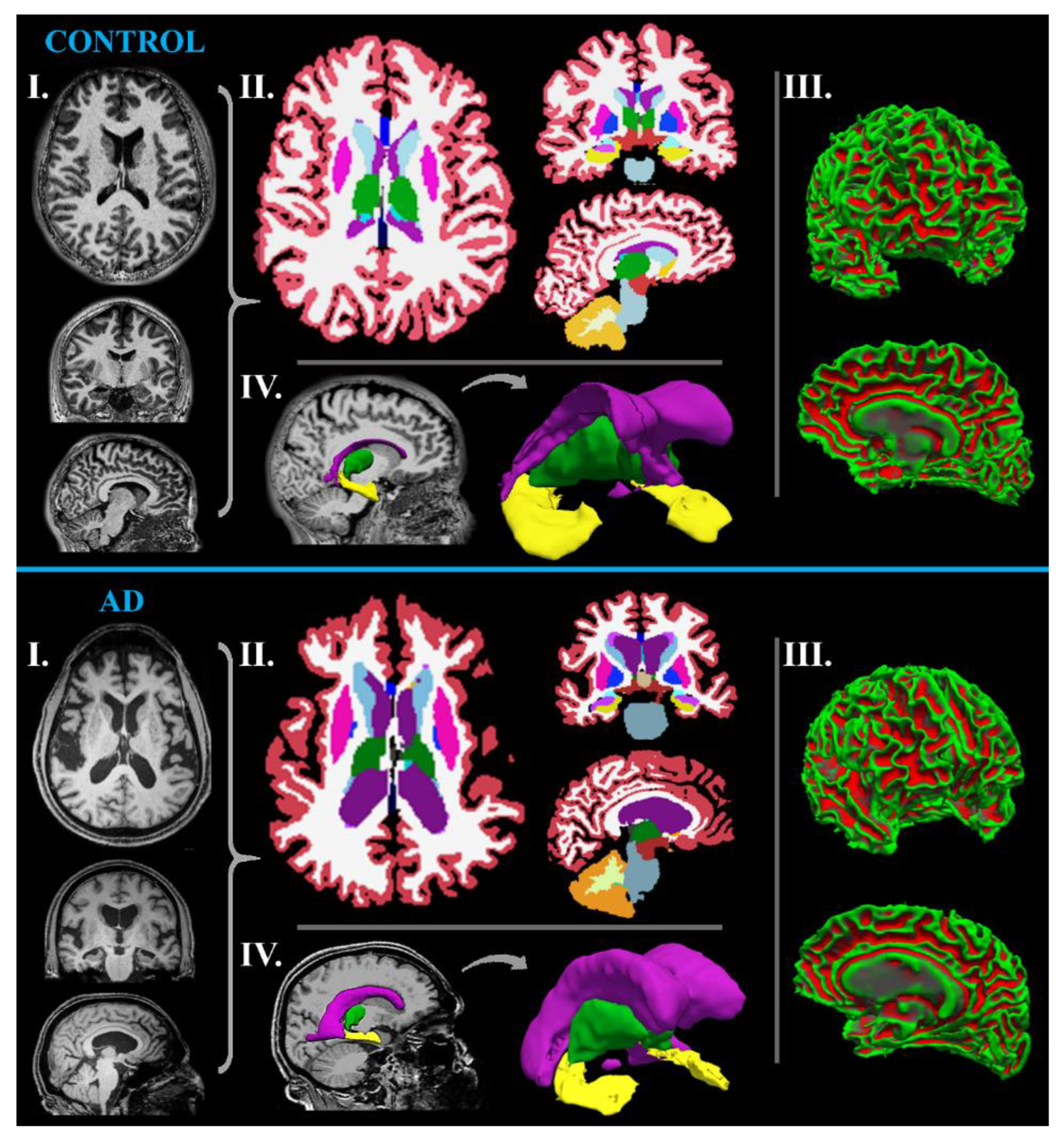
MR-Volumetry
fMRI
Perfusion MRI (ASL, DSC, and DCE)
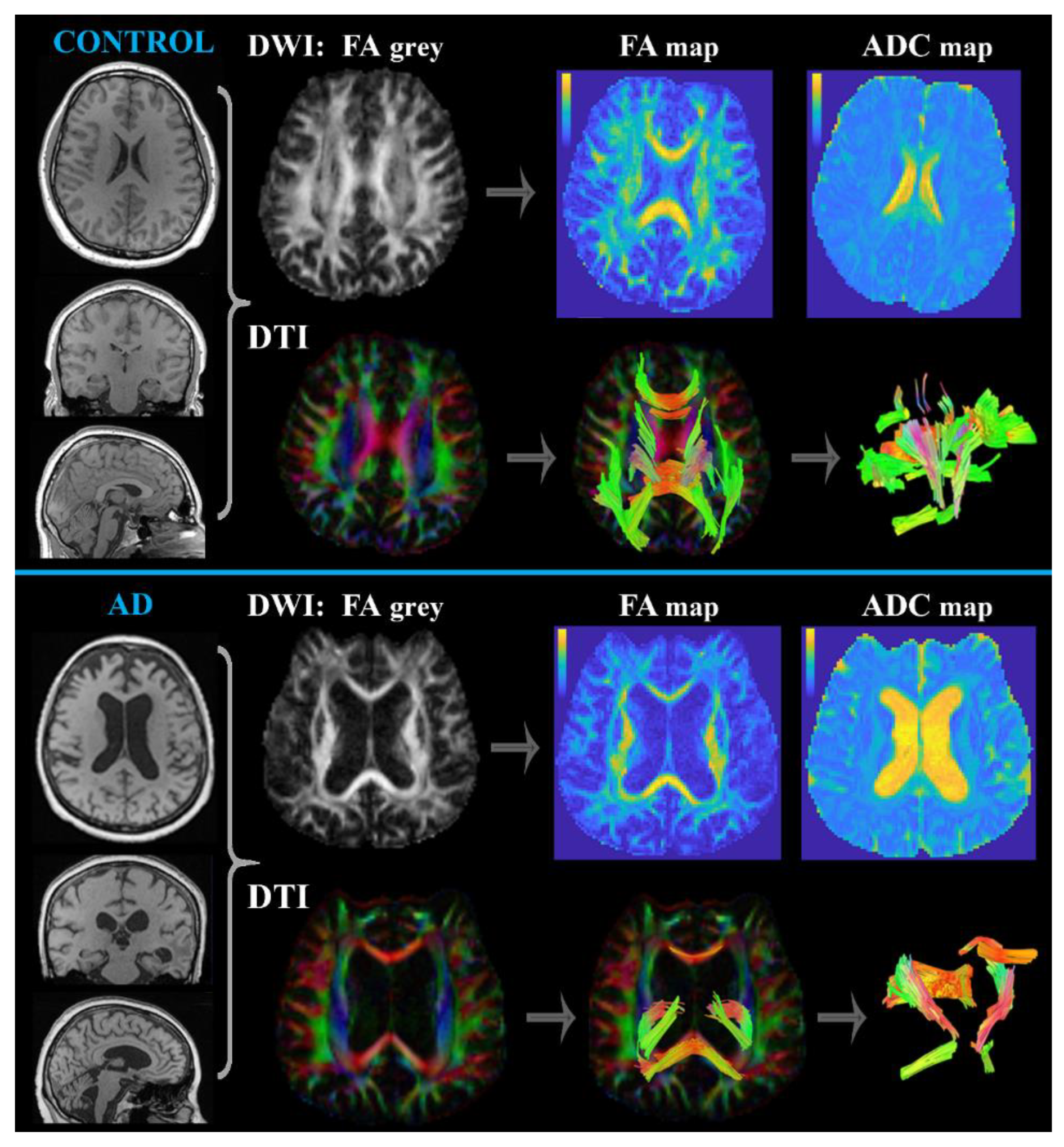
Diffusion MRI (DWI and DTI)
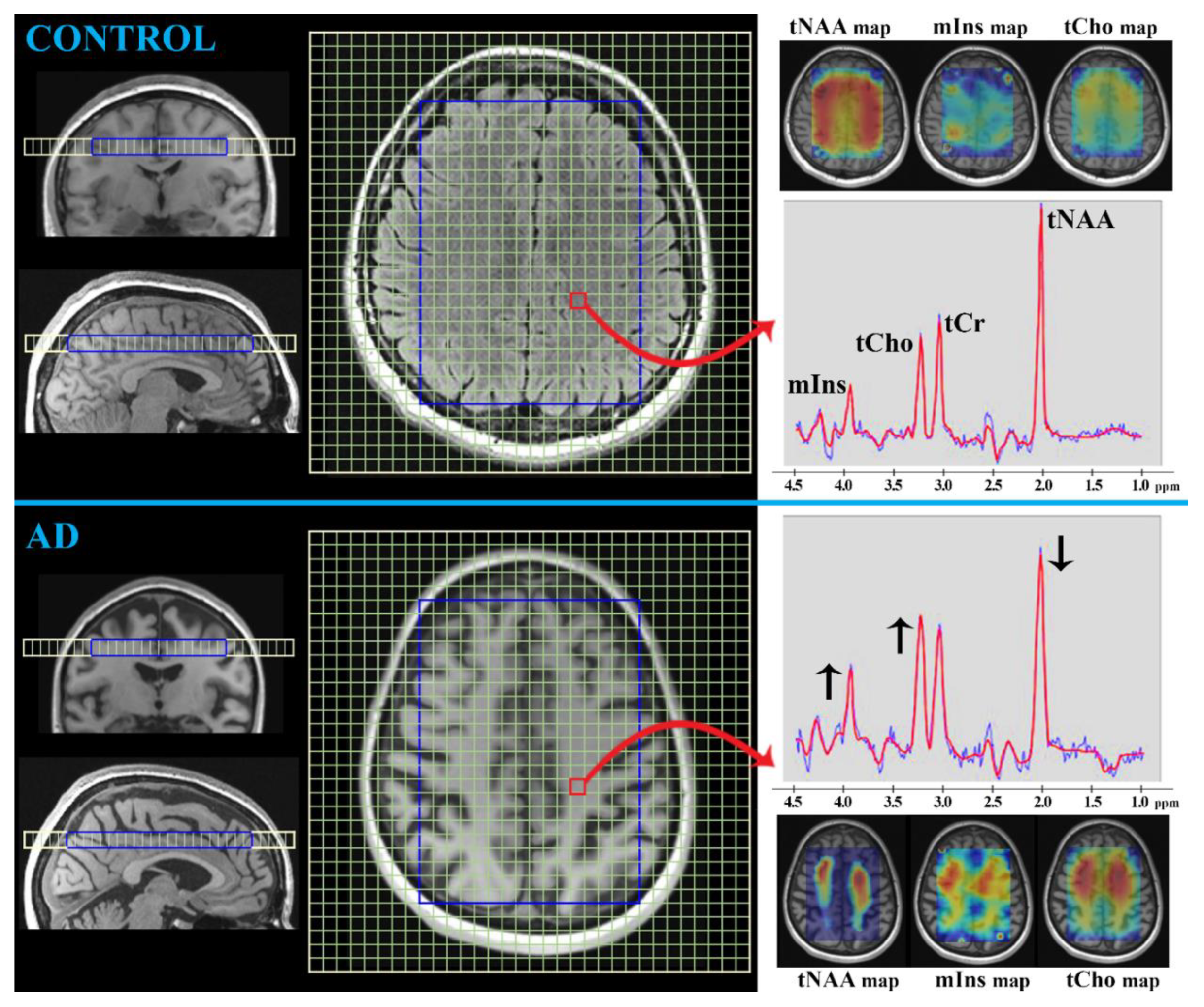
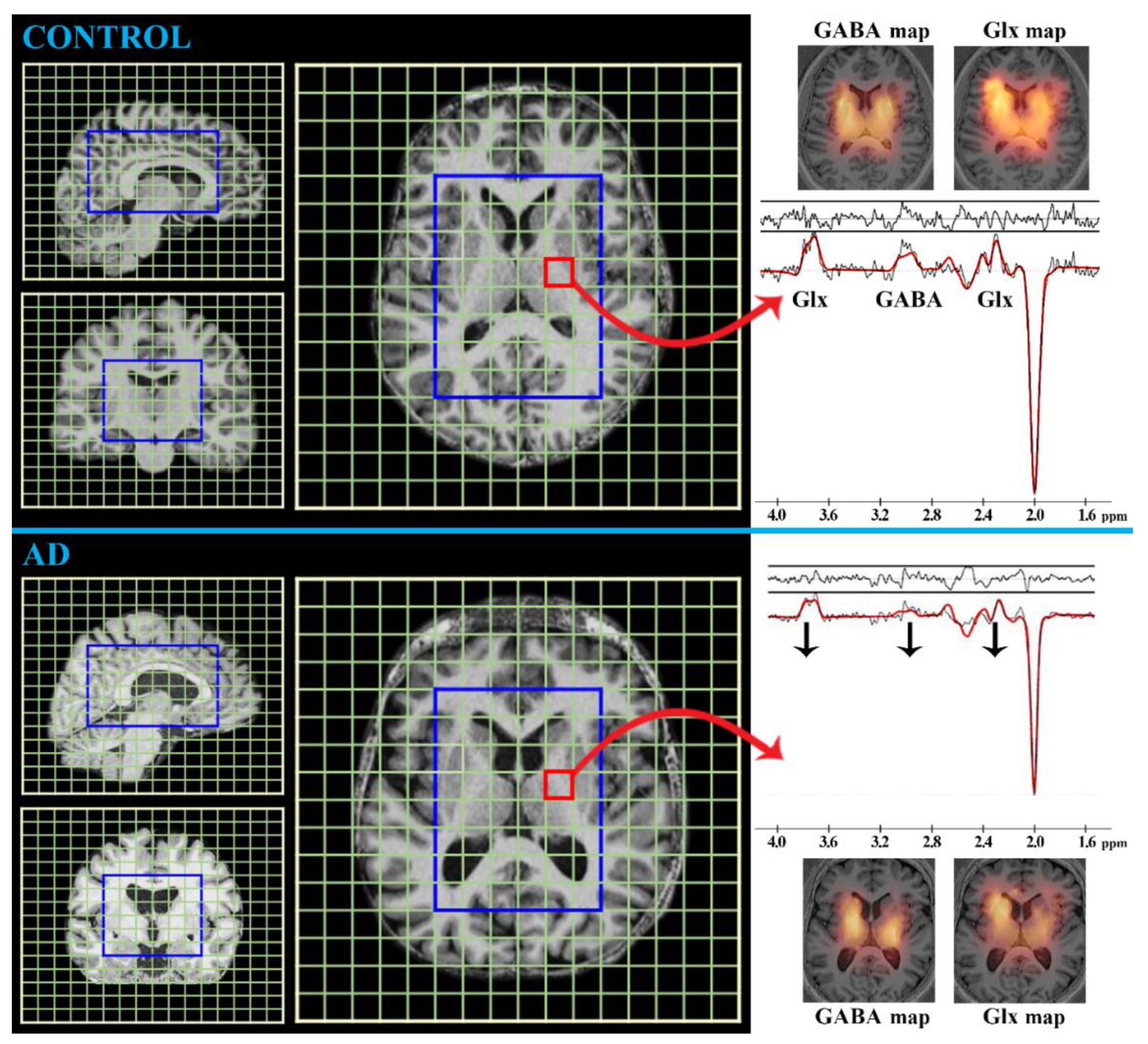
MRS
- ❖
- tCr (duplet at 3.0 ppm and 3.9 ppm; concentration ~6 mmol/L in the brain [20])
- ❖
- tNAA (singlet at 2.0 ppm; concentration ~15 mmol/L in the brain [20])
- ❖
- tCho (singlet at 3.2 ppm; concentration ~2 mmol/L in the brain [20])
- ❖
- mIns (multiples at 3.3, 3.5, and 4.0 ppm; concentration ~7 mmol/L in the brain [20])
- ❖
- Glx (multiples at 2.1, 2.4, and 3.7 ppm; concentration ~10 mmol/L in the brain [20])
- ❖
- GABA (multiples at 1.9, 2.3, and 3.0 ppm; concentration ~2 mmol/L in the brain [20])
- ❖
- GSH (multiples at 2.9 and 4.5 ppm; concentration ~1.5 mmol/L in the brain [86])
2.2. Nuclear Medicine Neuroimaging
2.2.1. PET Neuroimaging
18F-FDG PET
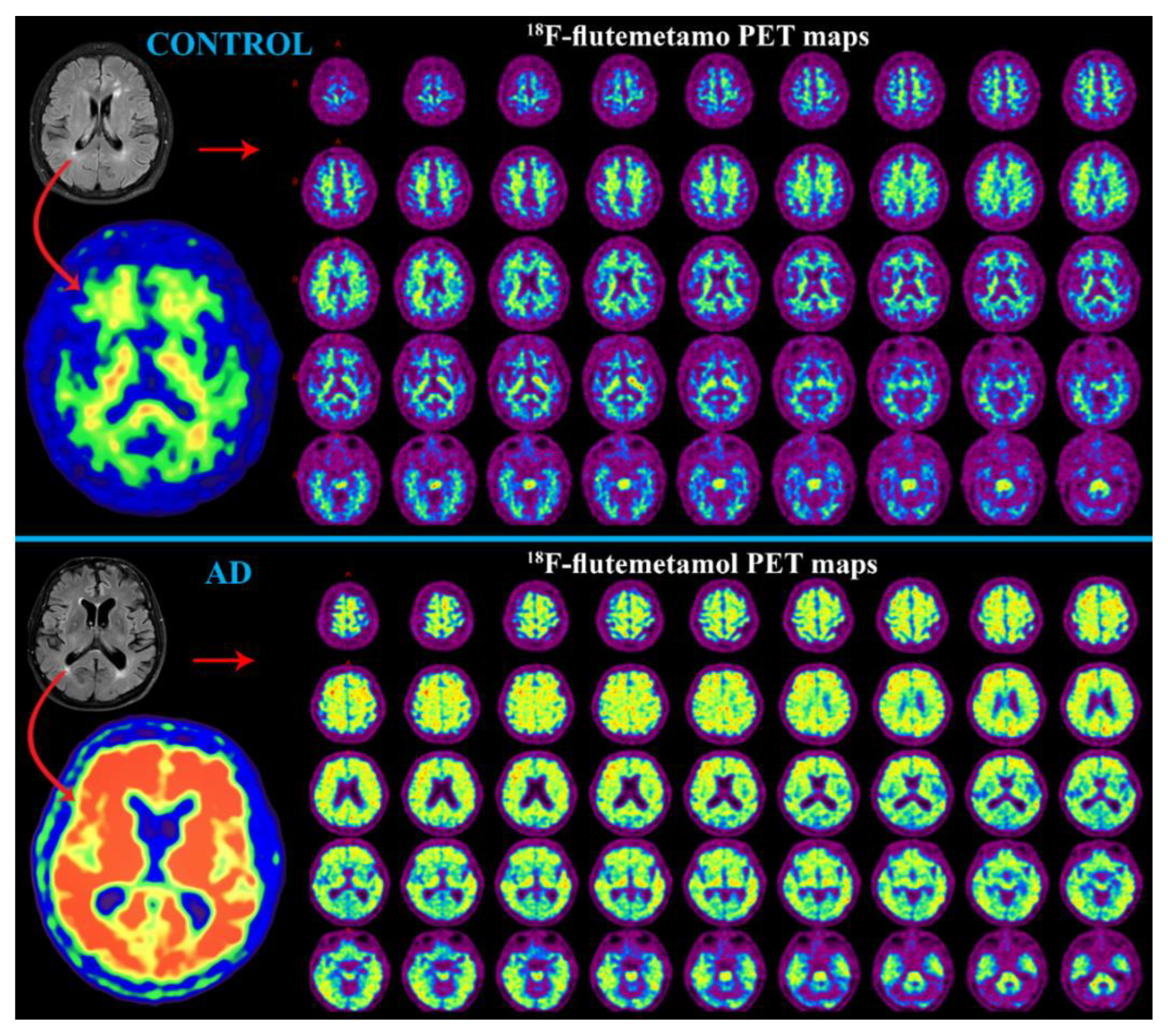
PET with Aβ Radiotracer
PET with Tau Radiotracer
2.2.2. SPECT Neuroimaging
SPECT Perfusion
SPECT with Aβ Radiotracer
2.3. Advantages, Disadvantages, and New trends in Neuroimaging of AD
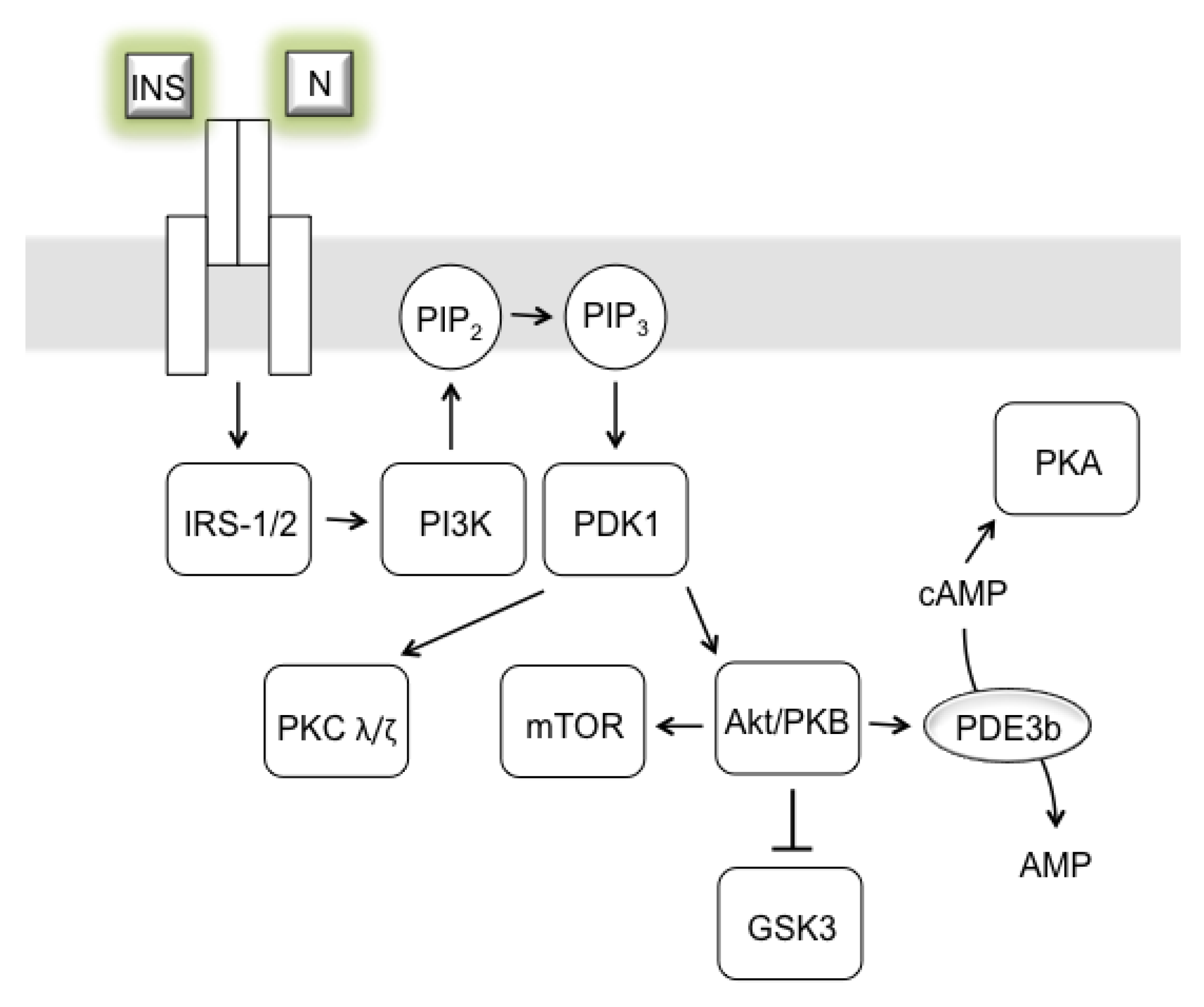
3. AD as Insulinopathy
3.1. Suggestive non-Neuronal Imaging in AD
3.1.1. Liver Imaging Linked with AD
3.1.2. Pancreas Imaging Linked with AD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beshir, S.A.; Aadithsoorya, A.M.; Parveen, A.; Goh, S.S.L.; Hussain, N.; Menon, V.B. Aducanumab Therapy to Treat Alzheimer’s Disease: A Narrative Review. Int. J. Alzheimers Dis. 2022, 2022, 9343514. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Rowley, P.A.; Samsonov, A.A.; Betthauser, T.J.; Pirasteh, A.; Johnson, S.C.; Eisenmenger, L.B. Amyloid and Tau PET Imaging of Alzheimer Disease and Other Neurodegenerative Conditions. Semin. Ultrasound CT MR 2020, 41, 572–583. [Google Scholar] [CrossRef]
- Patel, K.P.; Wymer, D.T.; Bhatia, V.K.; Duara, R.; Rajadhyaksha, C.D. Multimodality Imaging of Dementia: Clinical Importance and Role of Integrated Anatomic and Molecular Imaging. Radiographics 2020, 40, 200–222. [Google Scholar] [CrossRef]
- Valotassiou, V.; Malamitsi, J.; Papatriantafyllou, J.; Dardiotis, E.; Tsougos, I.; Psimadas, D.; Alexiou, S.; Hadjigeorgiou, G.; Georgoulias, P. SPECT and PET imaging in Alzheimer’s disease. Ann. Nucl. Med. 2018, 32, 583–593. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Giau, V.V.; Vo, T.K. Current advances in transdermal delivery of drugs for Alzheimer’s disease. Indian J. Pharmacol. 2017, 49, 145–154. [Google Scholar] [CrossRef]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front Biosci. (Elite Ed.) 2013, 5, 167–177. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Blazquez, E.; Velazquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol. (Lausanne) 2014, 5, 161. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin resistance in Alzheimer’s disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim. Biophys. Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Ferrando, R.; Damian, A. Brain SPECT as a Biomarker of Neurodegeneration in Dementia in the Era of Molecular Imaging: Still a Valid Option? Front Neurol. 2021, 12, 629442. [Google Scholar] [CrossRef]
- Mueller, S.G.; Schuff, N.; Weiner, M.W. Evaluation of treatment effects in Alzheimer’s and other neurodegenerative diseases by MRI and MRS. NMR Biomed. 2006, 19, 655–668. [Google Scholar] [CrossRef]
- Hnilicova, P.; Strbak, O.; Kolisek, M.; Kurca, E.; Zelenak, K.; Sivak, S.; Kantorova, E. Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 6117. [Google Scholar] [CrossRef]
- Zhang, Y.; Londos, E.; Minthon, L.; Wattmo, C.; Liu, H.; Aspelin, P.; Wahlund, L.O. Usefulness of computed tomography linear measurements in diagnosing Alzheimer’s disease. Acta Radiol. 2008, 49, 91–97. [Google Scholar] [CrossRef]
- Masters, C.L.; Cappai, R.; Barnham, K.J.; Villemagne, V.L. Molecular mechanisms for Alzheimer’s disease: Implications for neuroimaging and therapeutics. J. Neurochem. 2006, 97, 1700–1725. [Google Scholar] [CrossRef]
- Chandra, A.; Dervenoulas, G.; Politis, M.; Alzheimer’s Disease Neuroimaging, I. Magnetic resonance imaging in Alzheimer’s disease and mild cognitive impairment. J. Neurol. 2019, 266, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Berlow, Y.A.; Wells, W.M.; Ellison, J.M.; Sung, Y.H.; Renshaw, P.F.; Harper, D.G. Neuropsychiatric correlates of white matter hyperintensities in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2010, 25, 780–788. [Google Scholar] [CrossRef]
- Leocadi, M.; Canu, E.; Calderaro, D.; Corbetta, D.; Filippi, M.; Agosta, F. An update on magnetic resonance imaging markers in AD. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420947986. [Google Scholar] [CrossRef]
- Miller-Thomas, M.M.; Sipe, A.L.; Benzinger, T.L.; McConathy, J.; Connolly, S.; Schwetye, K.E. Multimodality Review of Amyloid-related Diseases of the Central Nervous System. Radiographics 2016, 36, 1147–1163. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Liu, R.; Qin, R.; Zhao, H.; Xu, Y. The associated volumes of sub-cortical structures and cognitive domain in patients of Mild Cognitive Impairment. J. Clin. Neurosci. 2018, 56, 56–62. [Google Scholar] [CrossRef]
- Sperling, R. Potential of functional MRI as a biomarker in early Alzheimer’s disease. Neurobiol. Aging 2011, 32, S37–S43. [Google Scholar] [CrossRef]
- Yamasaki, T.; Muranaka, H.; Kaseda, Y.; Mimori, Y.; Tobimatsu, S. Understanding the Pathophysiology of Alzheimer’s Disease and Mild Cognitive Impairment: A Mini Review on fMRI and ERP Studies. Neurol. Res. Int. 2012, 2012, 719056. [Google Scholar] [CrossRef]
- Glover, G.H. Overview of functional magnetic resonance imaging. Neurosurg. Clin. N. Am. 2011, 22, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Yang, H.; Zhang, J. Donepezil’s Effects on Brain Functions of Patients With Alzheimer Disease: A Regional Homogeneity Study Based on Resting-State Functional Magnetic Resonance Imaging. Clin. Neuropharmacol. 2019, 42, 42–48. [Google Scholar] [CrossRef]
- Wierenga, C.E.; Hays, C.C.; Zlatar, Z.Z. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, S411–S419. [Google Scholar] [CrossRef]
- Wolk, D.A.; Detre, J.A. Arterial spin labeling MRI: An emerging biomarker for Alzheimer’s disease and other neurodegenerative conditions. Curr. Opin. Neurol. 2012, 25, 421–428. [Google Scholar] [CrossRef]
- Essig, M.; Shiroishi, M.S.; Nguyen, T.B.; Saake, M.; Provenzale, J.M.; Enterline, D.; Anzalone, N.; Dorfler, A.; Rovira, A.; Wintermark, M.; et al. Perfusion MRI: The five most frequently asked technical questions. AJR Am. J. Roentgenol. 2013, 200, 24–34. [Google Scholar] [CrossRef]
- Asllani, I.; Habeck, C.; Scarmeas, N.; Borogovac, A.; Brown, T.R.; Stern, Y. Multivariate and univariate analysis of continuous arterial spin labeling perfusion MRI in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2008, 28, 725–736. [Google Scholar] [CrossRef]
- Dai, W.; Lopez, O.L.; Carmichael, O.T.; Becker, J.T.; Kuller, L.H.; Gach, H.M. Mild cognitive impairment and alzheimer disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 2009, 250, 856–866. [Google Scholar] [CrossRef]
- Binnewijzend, M.A.; Kuijer, J.P.; van der Flier, W.M.; Benedictus, M.R.; Moller, C.M.; Pijnenburg, Y.A.; Lemstra, A.W.; Prins, N.D.; Wattjes, M.P.; van Berckel, B.N.; et al. Distinct perfusion patterns in Alzheimer’s disease, frontotemporal dementia and dementia with Lewy bodies. Eur. Radiol. 2014, 24, 2326–2333. [Google Scholar] [CrossRef]
- Schuff, N.; Matsumoto, S.; Kmiecik, J.; Studholme, C.; Du, A.; Ezekiel, F.; Miller, B.L.; Kramer, J.H.; Jagust, W.J.; Chui, H.C.; et al. Cerebral blood flow in ischemic vascular dementia and Alzheimer’s disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimers Dement 2009, 5, 454–462. [Google Scholar] [CrossRef]
- Drake-Perez, M.; Boto, J.; Fitsiori, A.; Lovblad, K.; Vargas, M.I. Clinical applications of diffusion weighted imaging in neuroradiology. Insights Imaging 2018, 9, 535–547. [Google Scholar] [CrossRef]
- Gaddamanugu, S.; Shafaat, O.; Sotoudeh, H.; Sarrami, A.H.; Rezaei, A.; Saadatpour, Z.; Singhal, A. Clinical applications of diffusion-weighted sequence in brain imaging: Beyond stroke. Neuroradiology 2022, 64, 15–30. [Google Scholar] [CrossRef]
- Harrison, J.R.; Bhatia, S.; Tan, Z.X.; Mirza-Davies, A.; Benkert, H.; Tax, C.M.W.; Jones, D.K. Imaging Alzheimer’s genetic risk using diffusion MRI: A systematic review. Neuroimage Clin. 2020, 27, 102359. [Google Scholar] [CrossRef] [PubMed]
- Baliyan, V.; Das, C.J.; Sharma, R.; Gupta, A.K. Diffusion weighted imaging: Technique and applications. World J. Radiol. 2016, 8, 785–798. [Google Scholar] [CrossRef]
- Huisman, T.A. Diffusion-weighted and diffusion tensor imaging of the brain, made easy. Cancer Imaging 2010, 10, S163–S171. [Google Scholar] [CrossRef] [PubMed]
- Perea, R.D.; Rabin, J.S.; Fujiyoshi, M.G.; Neal, T.E.; Smith, E.E.; Van Dijk, K.R.A.; Hedden, T. Connectome-derived diffusion characteristics of the fornix in Alzheimer’s disease. Neuroimage Clin. 2018, 19, 331–342. [Google Scholar] [CrossRef]
- Soares, D.P.; Law, M. Magnetic resonance spectroscopy of the brain: Review of metabolites and clinical applications. Clin. Radiol. 2009, 64, 12–21. [Google Scholar] [CrossRef]
- Zhu, H.; Barker, P.B. MR spectroscopy and spectroscopic imaging of the brain. Methods Mol. Biol. 2011, 711, 203–226. [Google Scholar] [CrossRef]
- Gao, F.; Barker, P.B. Various MRS application tools for Alzheimer disease and mild cognitive impairment. AJNR Am. J. Neuroradiol. 2014, 35, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Maul, S.; Giegling, I.; Rujescu, D. Proton Magnetic Resonance Spectroscopy in Common Dementias-Current Status and Perspectives. Front Psychiatry 2020, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.F.; Backes, W.H.; Nicolay, K.; Kooi, M.E. 1H MR spectroscopy of the brain: Absolute quantification of metabolites. Radiology 2006, 240, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Edden, R.A.; Gao, F.; Wang, G.; Wu, L.; Zhao, B.; Wang, M.; Chan, Q.; Chen, W.; Barker, P.B. Decreased gamma-aminobutyric acid levels in the parietal region of patients with Alzheimer’s disease. J. Magn. Reason. Imaging 2015, 41, 1326–1331. [Google Scholar] [CrossRef] [Green Version]
- Tumati, S.; Martens, S.; Aleman, A. Magnetic resonance spectroscopy in mild cognitive impairment: Systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2013, 37, 2571–2586. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Kantarci, K. Magnetic resonance spectroscopy in Alzheimer’s disease. Neuropsychiatr Dis. Treat 2013, 9, 687–696. [Google Scholar] [CrossRef]
- Kantarci, K.; Weigand, S.D.; Petersen, R.C.; Boeve, B.F.; Knopman, D.S.; Gunter, J.; Reyes, D.; Shiung, M.; O’Brien, P.C.; Smith, G.E.; et al. Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tan, L.; Wang, H.F.; Liu, Y.; Yin, R.H.; Wang, W.Y.; Chang, X.L.; Jiang, T.; Yu, J.T. Magnetic Resonance Spectroscopy in Alzheimer’s Disease: Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2015, 46, 1049–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Shi, H.; Shen, Z.; Hou, L.; Luo, X.; Chen, X.; Liu, S.; Zhang, Y.; Zheng, D.; Tan, Y.; et al. 1H-proton magnetic resonance spectroscopy differentiates dementia with Lewy bodies from Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 953–966. [Google Scholar] [CrossRef]
- Zhu, X.; Schuff, N.; Kornak, J.; Soher, B.; Yaffe, K.; Kramer, J.H.; Ezekiel, F.; Miller, B.L.; Jagust, W.J.; Weiner, M.W. Effects of Alzheimer disease on fronto-parietal brain N-acetyl aspartate and myo-inositol using magnetic resonance spectroscopic imaging. Alzheimer Dis. Assoc Disord 2006, 20, 77–85. [Google Scholar] [CrossRef]
- Dixon, R.M.; Bradley, K.M.; Budge, M.M.; Styles, P.; Smith, A.D. Longitudinal quantitative proton magnetic resonance spectroscopy of the hippocampus in Alzheimer’s disease. Brain 2002, 125, 2332–2341. [Google Scholar] [CrossRef]
- Hancu, I.; Zimmerman, E.A.; Sailasuta, N.; Hurd, R.E. 1H MR spectroscopy using TE averaged PRESS: A more sensitive technique to detect neurodegeneration associated with Alzheimer’s disease. Magn. Reson Med. 2005, 53, 777–782. [Google Scholar] [CrossRef]
- Falini, A.; Bozzali, M.; Magnani, G.; Pero, G.; Gambini, A.; Benedetti, B.; Mossini, R.; Franceschi, M.; Comi, G.; Scotti, G.; et al. A whole brain MR spectroscopy study from patients with Alzheimer’s disease and mild cognitive impairment. Neuroimage 2005, 26, 1159–1163. [Google Scholar] [CrossRef]
- Jessen, F.; Traeber, F.; Freymann, N.; Maier, W.; Schild, H.H.; Heun, R.; Block, W. A comparative study of the different N-acetylaspartate measures of the medial temporal lobe in Alzheimer’s disease. Dement Geriatr. Cogn. Disord 2005, 20, 178–183. [Google Scholar] [CrossRef]
- Watanabe, T.; Shiino, A.; Akiguchi, I. Absolute quantification in proton magnetic resonance spectroscopy is useful to differentiate amnesic mild cognitive impairment from Alzheimer’s disease and healthy aging. Dement Geriatr. Cogn. Disord 2010, 30, 71–77. [Google Scholar] [CrossRef]
- Modrego, P.J.; Fayed, N. Longitudinal magnetic resonance spectroscopy as marker of cognitive deterioration in mild cognitive impairment. Am. J. Alzheimers Dis. Other Demen. 2011, 26, 631–636. [Google Scholar] [CrossRef]
- Mihara, M.; Hattori, N.; Abe, K.; Sakoda, S.; Sawada, T. Magnetic resonance spectroscopic study of Alzheimer’s disease and frontotemporal dementia/Pick complex. Neuroreport 2006, 17, 413–416. [Google Scholar] [CrossRef]
- Murray, M.E.; Przybelski, S.A.; Lesnick, T.G.; Liesinger, A.M.; Spychalla, A.; Zhang, B.; Gunter, J.L.; Parisi, J.E.; Boeve, B.F.; Knopman, D.S.; et al. Early Alzheimer’s disease neuropathology detected by proton MR spectroscopy. J. Neurosci. 2014, 34, 16247–16255. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Hasan, G. The InsP3 receptor: Its role in neuronal physiology and neurodegeneration. Bioessays 2005, 27, 1035–1047. [Google Scholar] [CrossRef]
- Waldman, A.D.; Rai, G.S. The relationship between cognitive impairment and in vivo metabolite ratios in patients with clinical Alzheimer’s disease and vascular dementia: A proton magnetic resonance spectroscopy study. Neuroradiology 2003, 45, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Shen, J. Glutamate. In Magnetic Resonance Spectroscopy; Charlotte Stagg, D.R., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 111–121. [Google Scholar]
- Hassel, B.; Dingledine, R. Glutamate and Glutamate Receptors. In Basic Neurochemistry; Scott, T., Brady George, J., Siegel, R., Albers, W., Donald, L., Eds.; Academic Press: Waltham, MA, USA, 2012; pp. 342–366. [Google Scholar]
- Azevedo, C.J.; Kornak, J.; Chu, P.; Sampat, M.; Okuda, D.T.; Cree, B.A.; Nelson, S.J.; Hauser, S.L.; Pelletier, D. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann. Neurol. 2014, 76, 269–278. [Google Scholar] [CrossRef]
- Hattori, N.; Abe, K.; Sakoda, S.; Sawada, T. Proton MR spectroscopic study at 3 Tesla on glutamate/glutamine in Alzheimer’s disease. Neuroreport 2002, 13, 183–186. [Google Scholar] [CrossRef]
- Huang, D.; Liu, D.; Yin, J.; Qian, T.; Shrestha, S.; Ni, H. Glutamate-glutamine and GABA in brain of normal aged and patients with cognitive impairment. Eur. Radiol. 2017, 27, 2698–2705. [Google Scholar] [CrossRef]
- Fayed, N.; Modrego, P.J.; Rojas-Salinas, G.; Aguilar, K. Brain glutamate levels are decreased in Alzheimer’s disease: A magnetic resonance spectroscopy study. Am. J. Alzheimers Dis. Other Demen. 2011, 26, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Riese, F.; Gietl, A.; Zolch, N.; Henning, A.; O’Gorman, R.; Kalin, A.M.; Leh, S.E.; Buck, A.; Warnock, G.; Edden, R.A.; et al. Posterior cingulate gamma-aminobutyric acid and glutamate/glutamine are reduced in amnestic mild cognitive impairment and are unrelated to amyloid deposition and apolipoprotein E genotype. Neurobiol. Aging 2015, 36, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Blamire, A.M.; Watson, R.; He, J.; Hayes, L.; O’Brien, J.T. Whole-brain patterns of (1)H-magnetic resonance spectroscopy imaging in Alzheimer’s disease and dementia with Lewy bodies. Transl. Psychiatry 2016, 6, e877. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U.; Bacher, R.; Vonbank, H.; Kemmler, G.; Lingg, A.; Marksteiner, J. Cognitive impairment: Assessment with brain magnetic resonance imaging and proton magnetic resonance spectroscopy. J. Clin. Psychiatry 2003, 64, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Reno, C.M.; Sharma, S.; Christensen, C.; Huang, Y.; Fisher, S.J. Insulin action in the brain regulates both central and peripheral functions. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E156–E163. [Google Scholar] [CrossRef]
- Murari, G.; Liang, D.R.; Ali, A.; Chan, F.; Mulder-Heijstra, M.; Verhoeff, N.; Herrmann, N.; Chen, J.J.; Mah, L. Prefrontal GABA Levels Correlate with Memory in Older Adults at High Risk for Alzheimer’s Disease. Cereb. Cortex Commun. 2020, 1, tgaa022. [Google Scholar] [CrossRef]
- Zhang, J.M. Human Brain Glutamate, Glutamine, γ-Aminobutyric Acid Proton Magnetic Resonance Spectral Quantification with the Fast Padé Transform. Master’s Thesis, University of California, Los Angeles, CA, USA, ProQuest Dissertations Publishing, Los Angeles, CA, USA, 2013. [Google Scholar]
- Puts, N.A.; Edden, R.A. In vivo magnetic resonance spectroscopy of GABA: A methodological review. Prog. Nucl. Magn. Reason. Spectrosc. 2012, 60, 29–41. [Google Scholar] [CrossRef]
- Canas, P.M.; Simoes, A.P.; Rodrigues, R.J.; Cunha, R.A. Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 2014, 76, 51–56. [Google Scholar] [CrossRef]
- Nilsen, L.H.; Melo, T.M.; Saether, O.; Witter, M.P.; Sonnewald, U. Altered neurochemical profile in the McGill-R-Thy1-APP rat model of Alzheimer’s disease: A longitudinal in vivo 1 H MRS study. J. Neurochem. 2012, 123, 532–541. [Google Scholar] [CrossRef]
- Gao, F.; Edden, R.A.; Li, M.; Puts, N.A.; Wang, G.; Liu, C.; Zhao, B.; Wang, H.; Bai, X.; Zhao, C.; et al. Edited magnetic resonance spectroscopy detects an age-related decline in brain GABA levels. Neuroimage 2013, 78, 75–82. [Google Scholar] [CrossRef]
- Jimenez-Balado, J.; Ycaza Herrera, A.; Igwe, K.; Klem, L.; Buyukturkoglu, K.; Irimia, A.; Chen, L.; Guo, J.; Brickman, A.M.; Eich, T.S. Reduced Hippocampal GABA+ Is Associated With Poorer Episodic Memory in Healthy Older Women: A Pilot Study. Front Behav. Neurosci. 2021, 15, 695416. [Google Scholar] [CrossRef]
- Jimenez-Balado, J.; Eich, T.S. GABAergic dysfunction, neural network hyperactivity and memory impairments in human aging and Alzheimer’s disease. Semin Cell Dev. Biol. 2021, 116, 146–159. [Google Scholar] [CrossRef]
- Murley, A.G.; Rouse, M.A.; Jones, P.S.; Ye, R.; Hezemans, F.H.; O’Callaghan, C.; Frangou, P.; Kourtzi, Z.; Rua, C.; Carpenter, T.A.; et al. GABA and glutamate deficits from frontotemporal lobar degeneration are associated with disinhibition. Brain 2020, 143, 3449–3462. [Google Scholar] [CrossRef]
- Bottino, F.; Lucignani, M.; Napolitano, A.; Dellepiane, F.; Visconti, E.; Rossi Espagnet, M.C.; Pasquini, L. In Vivo Brain GSH: MRS Methods and Clinical Applications. Antioxidants 2021, 10, 1407. [Google Scholar] [CrossRef]
- Mandal, P.K.; Shukla, D.; Tripathi, M.; Ersland, L. Cognitive Improvement with Glutathione Supplement in Alzheimer’s Disease: A Way Forward. J. Alzheimers Dis. 2019, 68, 531–535. [Google Scholar] [CrossRef]
- Terpstra, M.; Henry, P.G.; Gruetter, R. Measurement of reduced glutathione (GSH) in human brain using LCModel analysis of difference-edited spectra. Magn. Reson. Med. 2003, 50, 19–23. [Google Scholar] [CrossRef]
- Chiang, G.C.; Mao, X.; Kang, G.; Chang, E.; Pandya, S.; Vallabhajosula, S.; Isaacson, R.; Ravdin, L.D.; Alzheimer’s Disease Neuroimaging, I.; Shungu, D.C. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol. 2017, 38, 1130–1137. [Google Scholar] [CrossRef]
- Shukla, D.; Mandal, P.K.; Ersland, L.; Gruner, E.R.; Tripathi, M.; Raghunathan, P.; Sharma, A.; Chaithya, G.R.; Punjabi, K.; Splaine, C. A Multi-Center Study on Human Brain Glutathione Conformation using Magnetic Resonance Spectroscopy. J. Alzheimers Dis. 2018, 66, 517–532. [Google Scholar] [CrossRef]
- Saharan, S.; Mandal, P.K. The emerging role of glutathione in Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels—a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Zhang, C.; Rodriguez, C.; Spaulding, J.; Aw, T.Y.; Feng, J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Crisan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.G.; Andries, G.; Cainap, C.; Chis, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef]
- Lu, F.M.; Yuan, Z. PET/SPECT molecular imaging in clinical neuroscience: Recent advances in the investigation of CNS diseases. Quant Imaging Med. Surg 2015, 5, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Heurling, K.; Johansson, E.; Leuzy, A. Disturbances in brain energy metabolism in insulin resistance and diabetes and Alzheimer’s disease—Learnings from brain imaging biomarkers. Int. Rev. Neurobiol. 2020, 154, 111–130. [Google Scholar] [CrossRef]
- Yeo, J.M.; Lim, X.; Khan, Z.; Pal, S. Systematic review of the diagnostic utility of SPECT imaging in dementia. Eur. Arch Psychiatry Clin. Neurosci. 2013, 263, 539–552. [Google Scholar] [CrossRef]
- Lobotesis, K.; Fenwick, J.D.; Phipps, A.; Ryman, A.; Swann, A.; Ballard, C.; McKeith, I.G.; O’Brien, J.T. Occipital hypoperfusion on SPECT in dementia with Lewy bodies but not AD. Neurology 2001, 56, 643–649. [Google Scholar] [CrossRef]
- Driscoll, I.; Troncoso, J.C.; Rudow, G.; Sojkova, J.; Pletnikova, O.; Zhou, Y.; Kraut, M.A.; Ferrucci, L.; Mathis, C.A.; Klunk, W.E.; et al. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol. 2012, 124, 823–831. [Google Scholar] [CrossRef]
- Mathis, C.A.; Kuller, L.H.; Klunk, W.E.; Snitz, B.E.; Price, J.C.; Weissfeld, L.A.; Rosario, B.L.; Lopresti, B.J.; Saxton, J.A.; Aizenstein, H.J. In vivo assessment of amyloid-β deposition in nondemented very elderly subjects. Ann. Neurol. 2013, 73, 751–761. [Google Scholar] [CrossRef]
- Camus, V.; Payoux, P.; Barre, L.; Desgranges, B.; Voisin, T.; Tauber, C.; La Joie, R.; Tafani, M.; Hommet, C.; Chetelat, G.; et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Sperling, R.A.; Gidicsin, C.M.; Carmasin, J.S.; Maye, J.E.; Coleman, R.E.; Reiman, E.M.; Sabbagh, M.N.; Sadowsky, C.H.; Fleisher, A.S.; et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement 2013, 9, S72–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.F.; Rosenberg, P.B.; Zhou, Y.; Kumar, A.; Raymont, V.; Ravert, H.T.; Dannals, R.F.; Nandi, A.; Brasic, J.R.; Ye, W.; et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J. Nucl. Med. 2010, 51, 913–920. [Google Scholar] [CrossRef]
- Doraiswamy, P.M.; Sperling, R.A.; Coleman, R.E.; Johnson, K.A.; Reiman, E.M.; Davis, M.D.; Grundman, M.; Sabbagh, M.N.; Sadowsky, C.H.; Fleisher, A.S.; et al. Amyloid-beta assessed by florbetapir F 18 PET and 18-month cognitive decline: A multicenter study. Neurology 2012, 79, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Ong, K.; Mulligan, R.S.; Holl, G.; Pejoska, S.; Jones, G.; O’Keefe, G.; Ackerman, U.; Tochon-Danguy, H.; Chan, J.G.; et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J. Nucl. Med. 2011, 52, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Leuzy, A.; Gauthier, S.; Rosa-Neto, P. Developments in Tau PET Imaging. Can J. Neurol. Sci. 2014, 41, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.P.; Wardak, M.; Shao, W.; Dahlbom, M.; Kepe, V.; Liu, J.; Satyamurthy, N.; Small, G.W.; Barrio, J.R.; Huang, S.C. Quantitative analysis of [18F]FDDNP PET using subcortical white matter as reference region. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef]
- Mattsson, N.; Ossenkoppele, R.; Smith, R.; Strandberg, O.; Ohlsson, T.; Jogi, J.; Palmqvist, S.; Stomrud, E.; Hansson, O. Greater tau load and reduced cortical thickness in APOE epsilon4-negative Alzheimer’s disease: A cohort study. Alzheimers Res. Ther. 2018, 10, 77. [Google Scholar] [CrossRef]
- Van Audenhaege, K.; Van Holen, R.; Vandenberghe, S.; Vanhove, C.; Metzler, S.D.; Moore, S.C. Review of SPECT collimator selection, optimization, and fabrication for clinical and preclinical imaging. Med. Phys. 2015, 42, 4796–4813. [Google Scholar] [CrossRef]
- Sjogren, M.; Gustafson, L.; Wikkelso, C.; Wallin, A. Frontotemporal dementia can be distinguished from Alzheimer’s disease and subcortical white matter dementia by an anterior-to-posterior rCBF-SPET ratio. Dement Geriatr. Cogn. Disord 2000, 11, 275–285. [Google Scholar] [CrossRef]
- Davis, K.M.; Ryan, J.L.; Aaron, V.D.; Sims, J.B. PET and SPECT Imaging of the Brain: History, Technical Considerations, Applications, and Radiotracers. Semin. Ultrasound CT MR 2020, 41, 521–529. [Google Scholar] [CrossRef]
- Maya, Y.; Okumura, Y.; Kobayashi, R.; Onishi, T.; Shoyama, Y.; Barret, O.; Alagille, D.; Jennings, D.; Marek, K.; Seibyl, J.; et al. Preclinical properties and human in vivo assessment of 123I-ABC577 as a novel SPECT agent for imaging amyloid-beta. Brain 2016, 139, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Kulkarni, P.P.; Galatzer-Levy, I.R.; Bigio, B.; Nasca, C.; Zhang, Y. Modern views of machine learning for precision psychiatry. Patterns (NY) 2022, 3, 100602. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Huang, Z.; Sun, Z.; Zhang, Y.; Zhao, Q.; Cichocki, A.; Yang, Z.; Sole-Casals, J. Topological Network Analysis of Early Alzheimer’s Disease Based on Resting-State EEG. IEEE Trans Neural Syst. Rehabil. Eng. 2020, 28, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Zhang, L.; Shen, Y.L.; Chen, B. Interpretable Graph Convolutional Network Of Multi-Modality Brain Imaging For Alzheimer’s Disease Diagnosis. In Proceedings of the 2022 IEEE 19th International Symposium on Biomedical Imaging (ISBI), Kolkata, India, 28–31 March 2022; IEEE: Piscatvie, NJ, USA, 2022; pp. 1–5. [Google Scholar] [CrossRef]
- Desikan, R.S.; Cabral, H.J.; Hess, C.P.; Dillon, W.P.; Glastonbury, C.M.; Weiner, M.W.; Schmansky, N.J.; Greve, D.N.; Salat, D.H.; Buckner, R.L.; et al. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer’s disease. Brain 2009, 132, 2048–2057. [Google Scholar] [CrossRef]
- Rastenyte, D.; Matijosaitis, V.; Laucius, O.; Gleizniene, R.; Jesmanas, S.; Jureniene, K. Diagnostic Ability of Structural Transcranial Sonography in Patients with Alzheimer’s Disease. Diagnostics 2020, 10, 471. [Google Scholar] [CrossRef]
- Wearn, A.R.; Nurdal, V.; Saunders-Jennings, E.; Knight, M.J.; Isotalus, H.K.; Dillon, S.; Tsivos, D.; Kauppinen, R.A.; Coulthard, E.J. Measuring brain integrity using MRI: A novel biomarker for Alzheimer’s disease using T2 relaxometry. medRxiv 2020. medRxiv:2020.2005.2013.20100339. [Google Scholar] [CrossRef]
- Larsen, J.P.; Britt, W., 3rd; Kido, D.; Olson, B.L.; Holshouser, B.A.; Kirsch, W.M. Susceptibility-Weighted Magnetic Resonance Imaging in the Evaluation of Dementia. Radiol. Case Rep. 2007, 2, 102. [Google Scholar] [CrossRef]
- Haller, S.; Haacke, E.M.; Thurnher, M.M.; Barkhof, F. Susceptibility-weighted Imaging: Technical Essentials and Clinical Neurologic Applications. Radiology 2021, 299, 3–26. [Google Scholar] [CrossRef]
- Agostinone, J.; Alarcon-Martinez, L.; Gamlin, C.; Yu, W.Q.; Wong, R.O.L.; Di Polo, A. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain 2018, 141, 1963–1980. [Google Scholar] [CrossRef]
- Sponder, G.; Abdulhanan, N.; Frohlich, N.; Mastrototaro, L.; Aschenbach, J.R.; Rontgen, M.; Pilchova, I.; Cibulka, M.; Racay, P.; Kolisek, M. Overexpression of Na(+)/Mg(2+) exchanger SLC41A1 attenuates pro-survival signaling. Oncotarget 2018, 9, 5084–5104. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Pijet, B.; Pijet-Kucicka, M.; Gajewska, M.; Pajak, B.; Orzechowski, A. FOXO1 and GSK-3beta Are Main Targets of Insulin-Mediated Myogenesis in C2C12 Muscle Cells. PLoS ONE 2016, 11, e0146726. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Rask-Madsen, C.; Banks, W.A. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J. Physiol. 2018, 596, 4753–4765. [Google Scholar] [CrossRef]
- Csajbok, E.A.; Tamas, G. Cerebral cortex: A target and source of insulin? Diabetologia 2016, 59, 1609–1615. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern Med. Rev. 2009, 14, 373–379. [Google Scholar]
- Liu, X.; Teng, Z.; Cui, C.; Wang, R.; Liu, M.; Zhang, Y. Amyloid beta-derived diffusible ligands (ADDLs) induce abnormal expression of insulin receptors in rat hippocampal neurons. J. Mol. Neurosci. 2014, 52, 124–130. [Google Scholar] [CrossRef]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Lauer, A.A.; Mett, J.; Janitschke, D.; Thiel, A.; Stahlmann, C.P.; Bachmann, C.M.; Ritzmann, F.; Schrul, B.; Muller, U.C.; Stein, R.; et al. Regulatory feedback cycle of the insulin-degrading enzyme and the amyloid precursor protein intracellular domain: Implications for Alzheimer’s disease. Aging Cell 2020, 19, e13264. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Barbiellini Amidei, C.F.A.; Dumurgier, J.; Machado-Fragua, M.D.; Tabak, A.G.; van Sloten, T.; Kivimaki, M.; Dugravot, A.; Sabia, S.; Singh-Manoux, A. Association Between Age at Diabetes Onset and Subsequent Risk of Dementia. JAMA 2021, 325, 1640–1649. [Google Scholar] [CrossRef]
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Rensink, A.A.; Otte-Holler, I.; de Boer, R.; Bosch, R.R.; ten Donkelaar, H.J.; de Waal, R.M.; Verbeek, M.M.; Kremer, B. Insulin inhibits amyloid beta-induced cell death in cultured human brain pericytes. Neurobiol. Aging 2004, 25, 93–103. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Adams, K.L.; Gallo, V. The diversity and disparity of the glial scar. Nat. Neurosci. 2018, 21, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Guo, D.H.; Hernandez, C.M.; Stranahan, A.M. Endothelial Adora2a Activation Promotes Blood-Brain Barrier Breakdown and Cognitive Impairment in Mice with Diet-Induced Insulin Resistance. J. Neurosci. 2019, 39, 4179–4192. [Google Scholar] [CrossRef]
- Vandal, M.; Bourassa, P.; Calon, F. Can insulin signaling pathways be targeted to transport Abeta out of the brain? Front Aging Neurosci. 2015, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Khoramipour, K.; Chamari, K.; Hekmatikar, A.A.; Ziyaiyan, A.; Taherkhani, S.; Elguindy, N.M.; Bragazzi, N.L. Adiponectin: Structure, Physiological Functions, Role in Diseases, and Effects of Nutrition. Nutrients 2021, 13, 1180. [Google Scholar] [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Shabalala, S.C.; Dludla, P.V.; Mabasa, L.; Kappo, A.P.; Basson, A.K.; Pheiffer, C.; Johnson, R. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed. Pharmacother. 2020, 131, 110785. [Google Scholar] [CrossRef]
- Ng, R.C.; Cheng, O.Y.; Jian, M.; Kwan, J.S.; Ho, P.W.; Cheng, K.K.; Yeung, P.K.; Zhou, L.L.; Hoo, R.L.; Chung, S.K.; et al. Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener 2016, 11, 71. [Google Scholar] [CrossRef]
- Chopra, I.; Li, H.F.; Wang, H.; Webster, K.A. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 2012, 55, 783–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence. Biomed. Res. Int. 2020, 2020, 9895121. [Google Scholar] [CrossRef]
- Qian, K.Z.S.; Xie, K.; Yu, D.; Yang, R.; Gong, D.W. Hepatic ALT isoenzymes are elevated in gluconeogenic conditions including diabetes and suppressed by insulin at the protein level. Diabetes/Metab. Res. Rev. 2015, 31, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Serres, S.B.E.; Franconi, J.M.; Merle, M. Brain pyruvate recycling and peripheral metabolism: An NMR analysis ex vivo of acetate and glucose metabolism in the rat. J. Neurochem. 2007, 101, 1428–1440. [Google Scholar] [CrossRef]
- Francis, P.T. Glutamatergic systems in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S15–S21. [Google Scholar] [CrossRef] [PubMed]
- Onaolapo, A.Y.; Onaolapo, O.J. Peripheral and Central Glutamate Dyshomeostasis in Neurodegenerative Disorders. Curr. Neuropharmacol. 2021, 19, 1069–1089. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Angeretti, N.; Lucca, E.; De Simoni, M.G.; Forloni, G. Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci. Lett. 1995, 188, 70–74. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Song, J. Novel insights into non-alcoholic fatty liver disease and dementia: Insulin resistance, hyperammonemia, gut dysbiosis, vascular impairment, and inflammation. Cell Biosci. 2022, 12, 99. [Google Scholar] [CrossRef]
- Kim, D.G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflammation. 2016, 13, 1. [Google Scholar] [CrossRef]
- Nho, K.; Kueider-Paisley, A.; Ahmad, S.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; et al. Association of Altered Liver Enzymes With Alzheimer Disease Diagnosis, Cognition, Neuroimaging Measures, and Cerebrospinal Fluid Biomarkers. JAMA Netw. Open 2019, 2, e197978. [Google Scholar] [CrossRef]
- Bril, F.; Cusi, K. Management of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes: A Call to Action. Diabetes Care 2017, 40, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Steven, S.; Tiniakos, D.; Taylor, R. Quantification of intrapancreatic fat in type 2 diabetes by MRI. PLoS ONE 2017, 12, e0174660. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Chau, W.; Callaway, M.P.; Dayan, C.M. Magnetic resonance imaging: A reliable method for measuring pancreatic volume in Type 1 diabetes. Diabet. Med. 2007, 24, 35–40. [Google Scholar] [CrossRef]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Sun, M.K.; Alkon, D.L. Links between Alzheimer’s disease and diabetes. Timely Top. Medicine. Cardiovasc. Dis. 2006, 10, E24. [Google Scholar] [CrossRef]
- Lin, L. Commonality between Diabetes and Alzheimer’s Disease and a New Strategy for the Therapy. Clin. Med. Pathol. 2008, 1, CPath-S667. [Google Scholar] [CrossRef]
- Kim, T.H.J.C.W.; Jun, H.Y.; Lee, C.; Noh, S.; Kim, J.E.; Kim, S.; Yoon, K.H. Accuracy of proton magnetic resonance for diagnosing non-alcoholic steatohepatitis: A meta-analysis. Sci. Rep. 2019, 9, 15002. [Google Scholar] [CrossRef]
- Liao, Y.Y.; Yang, K.C.; Lee, M.J.; Huang, K.C.; Chen, J.D.; Yeh, C.K. Multifeature analysis of an ultrasound quantitative diagnostic index for classifying nonalcoholic fatty liver disease. Sci. Rep. 2016, 6, 35083. [Google Scholar] [CrossRef]
- Pasanta, D.; Htun, K.T.; Pan, J.; Tungjai, M.; Kaewjaeng, S.; Kim, H.; Kaewkhao, J.; Kothan, S. Magnetic Resonance Spectroscopy of Hepatic Fat from Fundamental to Clinical Applications. Diagnostics 2021, 11, 842. [Google Scholar] [CrossRef]
- Schwenzer, N.F.; Springer, F.; Schraml, C.; Stefan, N.; Machann, J.; Schick, F. Non-invasive assessment and quantification of liver steatosis by ultrasound, computed tomography and magnetic resonance. J. Hepatol. 2009, 51, 433–445. [Google Scholar] [CrossRef]
- Naeem, M.; Bulow, R.; Schipf, S.; Werner, N.; Dorr, M.; Lerch, M.M.; Kuhn, J.P.; Rathmann, W.; Nauck, M.; Markus, M.R.P.; et al. Association of hepatic steatosis derived from ultrasound and quantitative MRI with prediabetes in the general population. Sci. Rep. 2021, 11, 13276. [Google Scholar] [CrossRef]
- Li, Z.; Sun, J.; Yang, X. Recent advances in molecular magnetic resonance imaging of liver fibrosis. Biomed. Res. Int. 2015, 2015, 595467. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.R.; Semelka, R.C. Magnetic resonance imaging of the liver: Review of techniques and approach to common diseases. Semin. Ultrasound CT MR 2005, 26, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Seyedpour, S.M.; Nabati, M.; Lambers, L.; Nafisi, S.; Tautenhahn, H.M.; Sack, I.; Reichenbach, J.R.; Ricken, T. Application of Magnetic Resonance Imaging in Liver Biomechanics: A Systematic Review. Front Physiol. 2021, 12, 733393. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Kim, S.M.; Kim, Y.S.; Kwon, O.S.; Shin, S.K.; Kim, K.K.; Lee, K.; Park, I.B.; Choi, C.S.; Chung, D.H.; et al. Magnetic Resonance-Based Assessments Better Capture Pathophysiologic Profiles and Progression in Nonalcoholic Fatty Liver Disease. Diabetes Metab. J. 2021, 45, 739–752. [Google Scholar] [CrossRef]
- Brancato, V.; Della Pepa, G.; Bozzetto, L.; Vitale, M.; Annuzzi, G.; Basso, L.; Cavaliere, C.; Salvatore, M.; Rivellese, A.A.; Monti, S. Evaluation of a Whole-Liver Dixon-Based MRI Approach for Quantification of Liver Fat in Patients with Type 2 Diabetes Treated with Two Isocaloric Different Diets. Diagnostics 2022, 12, 514. [Google Scholar] [CrossRef]
- Qayyum, A. MR spectroscopy of the liver: Principles and clinical applications. Radiographics 2009, 29, 1653–1664. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Zhao, J.N.; Zhong, W.J.; Luo, Y.D.; Wu, W.; Chen, W.J.; Dai, Y.B. The Value of Proton Magnetic Resonance Spectroscopy in High-Intensity Focused Ultrasound Treatment of Experimental Liver Cancer. Transl Oncol. 2015, 8, 163–168. [Google Scholar] [CrossRef]
- Burute, N.; Nisenbaum, R.; Jenkins, D.J.; Mirrahimi, A.; Anthwal, S.; Colak, E.; Kirpalani, A. Pancreas volume measurement in patients with Type 2 diabetes using magnetic resonance imaging-based planimetry. Pancreatology 2014, 14, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Virostko, J. Quantitative Magnetic Resonance Imaging of the Pancreas of Individuals With Diabetes. Front Endocrinol. (Lausanne) 2020, 11, 592349. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Kim, H.W.; Nayak, K.S.; Goran, M.I. Comparison of fat-water MRI and single-voxel MRS in the assessment of hepatic and pancreatic fat fractions in humans. Obesity (Silver Spring) 2010, 18, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Perman, W.H.; Balci, N.C.; Akduman, I. Review of magnetic resonance spectroscopy in the liver and the pancreas. Top Magn. Reson. Imaging 2009, 20, 89–97. [Google Scholar] [CrossRef]
- Cassidy, F.H.; Yokoo, T.; Aganovic, L.; Hanna, R.F.; Bydder, M.; Middleton, M.S.; Hamilton, G.; Chavez, A.D.; Schwimmer, J.B.; Sirlin, C.B. Fatty liver disease: MR imaging techniques for the detection and quantification of liver steatosis. Radiographics 2009, 29, 231–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Byun, J.H.; Park, B.J.; Park, S.H.; Kim, N.; Park, B.; Kim, J.K.; Lee, M.G. Quantitative analysis of diffusion-weighted magnetic resonance imaging of the pancreas: Usefulness in characterizing solid pancreatic masses. J. Magn. Reson. Imaging 2008, 28, 928–936. [Google Scholar] [CrossRef]
- Tirkes, T.; Fogel, E.L.; Sherman, S.; Lin, C.; Swensson, J.; Akisik, F.; Sandrasegaran, K. Detection of exocrine dysfunction by MRI in patients with early chronic pancreatitis. Abdom. Radiol. (NY) 2017, 42, 544–551. [Google Scholar] [CrossRef]
- Weis, J.; Ahlstrom, H.; Korsgren, O. Proton MR spectroscopy of human pancreas allografts. MAGMA 2019, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Principal Advantages | Principal Disadvantages |
|---|---|---|
| CT | short examination time; less critical contraindication criteria | X-ray exposure; contrast agents may worsen kidney function; poorer definition of critical brain structures (hippocampus, brain stem) |
| MR methods | no radiation exposure; less expensive examination; widespread availability; wide range of examinations; (standardly) no contrast agents | relatively poor pathological specificity; contraindication criteria (presence of magnetic non-compatible medical devices or foreign bodies); uncomfortable with noise and limited size of MR-gantry |
| MRI | high-resolution morphology | insensitive to small amount of calcification and bone fracture |
| MR-volumetry | sensitive tissue volume changes | several non-standardized softwares |
| fMRI | disrupted brain functions | challenging tasks for attention |
| MRS | non-invasive detection of metabolic changes | robustness; difficult absolute quantification; non-unambiguous predictive or distinguishable biomarkers |
| diffusion MRI (DWI, DTI) | demyelinated axons; damaged nerve tracks; 3D visualization of neural pathways | pitfall of mucinous or hemorrhagic lesions; highly motion sensitive |
| perfusion MRI (ASL, DCE, DSC) | acute inflammation; tissue degradation; ! in the case of ASL no contrast agent | susceptibility artifacts; ! in the case of non-ASL methods (DCE, DSC) contrast agent |
| PET and SPECT | definitive diagnostics; functional and molecular pathological processes | radiation exposure; more expensive examination; not routine equipment; Tau tracer (Tauvid©) not registered by EMA and thus not commercially available in EU |
| Methods | Utility | Principal Advantages | Principal Disadvantages |
|---|---|---|---|
| EEG [115,116] | neuronal electrical activity is measured via electrodes positioned on the scalp | low cost examination; easy and non-invasive performance | multiple signal distortions |
| ECoG [115] | intracranial EEG that measures signals directly from the cortical surface | excellent spatial and spectral resolution minimizing inter-electrode spacing | invasive performance; necessity of surgery |
| MEG [115,116] | EEG measured as magnetic fields produced by brain and evaluating by superconducting quantum interference devices | non-invasiveness; high temporal and spatial resolution | technically chalenging (necessity of specialized shielding to eliminate the magnetic interference) |
| TSC [119] | evaluating brain tissue echogenicity through an intact skull bone using ultrasound technique | non-invasiveness; low cost examination | insufficient temporal acoustic bone windows; diagnostic precision depends on neurosonographers skills |
| T2-relaxometry [20,120] | T2 relaxation time (one of MRI measurable parameters) is sensitive to microstructural changes (i.e., Aβ, Tau, and iron deposition; changes in water homeostasis) | non-invasiveness; early changes detection also in deep brain structures | sensitivity to magnetic susceptibilities, magnetic field variation, and inhomogeneities |
| SWI [121,122] | MRI method suitable for the detection of microhemorrhages that may indicate cerebral amyloid angiopathy | non-invasiveness; easy (routine) and fast examination of the whole brain | difficult to differentiate small venous structures from small hemorrhages and thrombosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hnilicova, P.; Kantorova, E.; Sutovsky, S.; Grofik, M.; Zelenak, K.; Kurca, E.; Zilka, N.; Parvanovova, P.; Kolisek, M. Imaging Methods Applicable in the Diagnostics of Alzheimer’s Disease, Considering the Involvement of Insulin Resistance. Int. J. Mol. Sci. 2023, 24, 3325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043325
Hnilicova P, Kantorova E, Sutovsky S, Grofik M, Zelenak K, Kurca E, Zilka N, Parvanovova P, Kolisek M. Imaging Methods Applicable in the Diagnostics of Alzheimer’s Disease, Considering the Involvement of Insulin Resistance. International Journal of Molecular Sciences. 2023; 24(4):3325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043325
Chicago/Turabian StyleHnilicova, Petra, Ema Kantorova, Stanislav Sutovsky, Milan Grofik, Kamil Zelenak, Egon Kurca, Norbert Zilka, Petra Parvanovova, and Martin Kolisek. 2023. "Imaging Methods Applicable in the Diagnostics of Alzheimer’s Disease, Considering the Involvement of Insulin Resistance" International Journal of Molecular Sciences 24, no. 4: 3325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043325