
Rapid Decline of Serum Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) in Non-Cirrhotic Patients with Chronic Hepatitis C Infection Receiving Direct-Acting Antiviral Therapy


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. PCSK9 ELISA
2.3. Statistical Analysis
3. Results
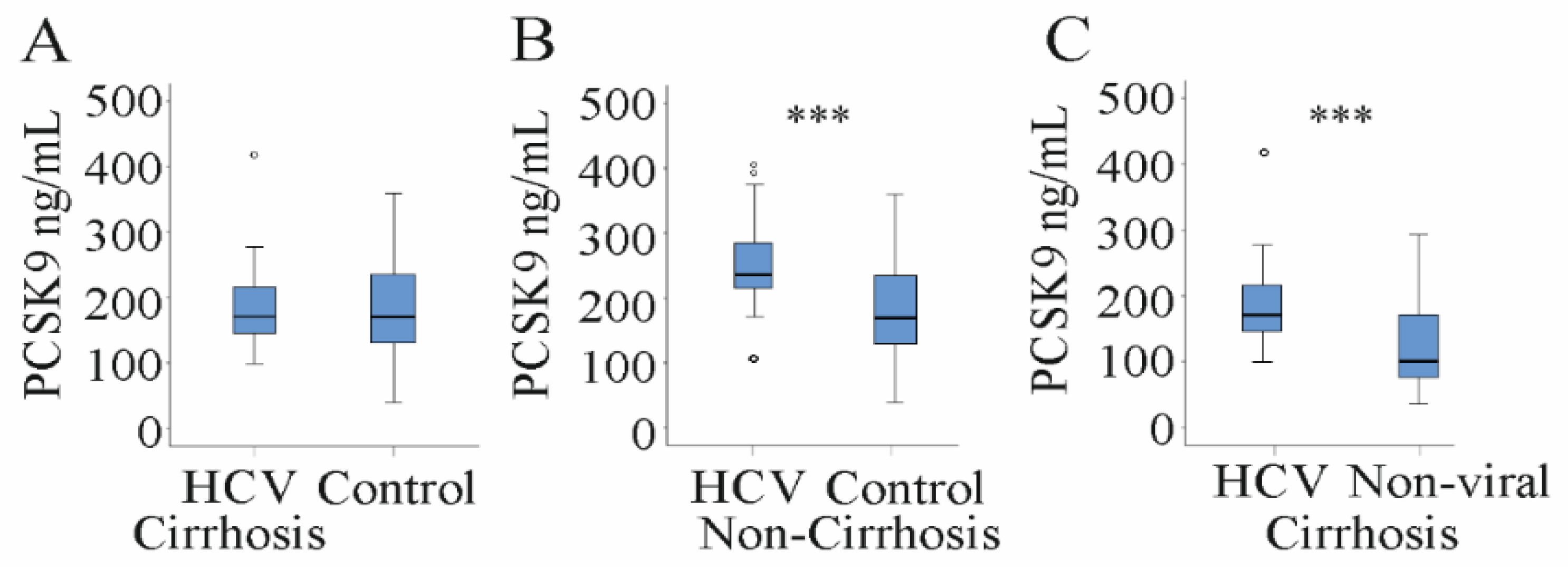
3.1. Serum PCSK9 Is Increased in HCV Patients
3.2. Serum PCSK9 Correlates with Markers of Liver Injury, Viral Load and LDL in HCV Patients
3.3. Serum PCSK9 in Relation to Liver Steatosis, Cirrhosis and Fibrosis Scores
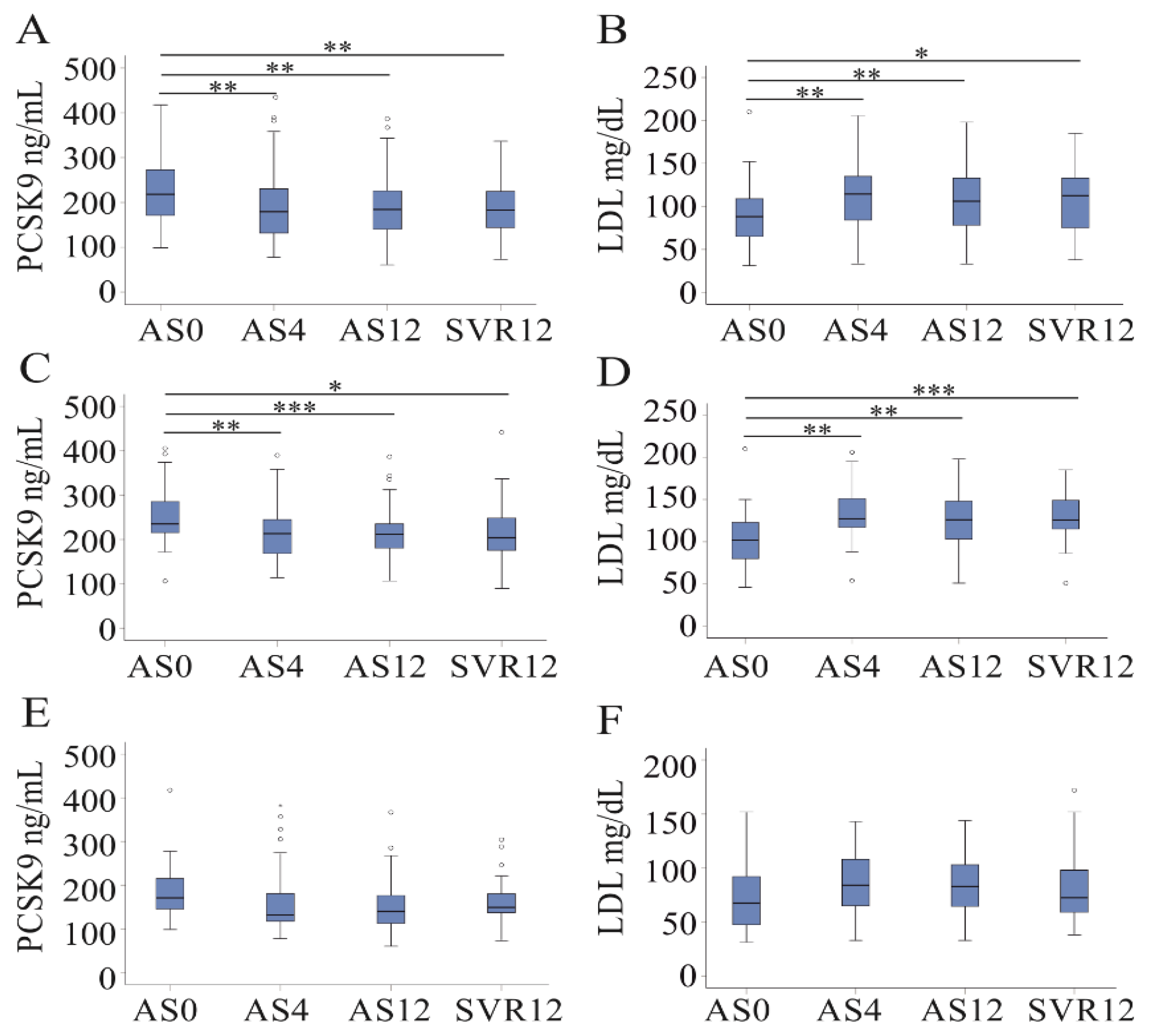
3.4. DAA Therapy Reduces Serum PCSK9 Levels in Non-Cirrhosis Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casiraghi, M.A.; De Paschale, M.; Romano, L.; Biffi, R.; Assi, A.; Binelli, G.; Zanetti, A.R. Long-term outcome (35 years) of hepatitis C after acquisition of infection through mini transfusions of blood given at birth. Hepatology 2004, 39, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, D.B.; Jiang, W.; Chen, X.B.; Xiao, G.B.; Wang, Y.H.; Wang, M.L.; Tao, Y.C.; Chen, E.Q. Efficacy and safety of sofosbuvir-based pangenotypic direct-acting antiviral agents for chronic hepatitis C patients without genotype determination: Real-world experience of a retrospective study. Medicine 2020, 99, e22726. [Google Scholar] [CrossRef]
- Nakajima, T.; Karino, Y.; Hige, S.; Suii, H.; Tatsumi, R.; Yamaguchi, M.; Arakawa, T.; Kuwata, Y.; Hasegawa, T.; Toyota, J. Factors affecting the recovery of hepatic reserve after sustained virologic response by direct-acting antiviral agents in chronic hepatitis C virus-infected patients. J. Gastroenterol. Hepatol. 2020, 36, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Peschel, G.; Grimm, J.; Gulow, K.; Muller, M.; Buechler, C.; Weigand, K. Chemerin Is a Valuable Biomarker in Patients with HCV Infection and Correlates with Liver Injury. Diagnostics 2020, 10, 974. [Google Scholar] [CrossRef] [PubMed]
- Hasan, Y.; Brown, K. Viral eradication restores normal iron status in chronic hepatitis C patients with abnormal iron studies. Ann. Hepatol. 2020, 19, 422–426. [Google Scholar] [CrossRef]
- Verna, E.C.; Morelli, G.; Terrault, N.A.; Lok, A.S.; Lim, J.K.; Di Bisceglie, A.M.; Zeuzem, S.; Landis, C.S.; Kwo, P.; Hassan, M.; et al. DAA therapy and long-term hepatic function in advanced/decompensated cirrhosis: Real-world experience from HCV-TARGET cohort. J. Hepatol. 2020, 73, 540–548. [Google Scholar] [CrossRef]
- Enomoto, M.; Kawada, N. The Moral of Hepatic Fibrosis: Don’t Always Believe Noninvasive Fibrosis Measurements. Dig. Dis. Sci. 2020, 65, 1293–1295. [Google Scholar] [CrossRef] [Green Version]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef] [Green Version]
- Feder, S.; Wiest, R.; Weiss, T.S.; Aslanidis, C.; Schacherer, D.; Krautbauer, S.; Liebisch, G.; Buechler, C. Proprotein convertase subtilisin/kexin type 9 (PCSK9) levels are not associated with severity of liver disease and are inversely related to cholesterol in a cohort of thirty eight patients with liver cirrhosis. Lipids Health Dis. 2021, 20, 1–14. [Google Scholar] [CrossRef]
- Asselah, T.; Rubbia-Brandt, L.; Marcellin, P.; Negro, F. Steatosis in chronic hepatitis C: Why does it really matter? Gut 2006, 55, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef]
- Hashimoto, S.; Yatsuhashi, H.; Abiru, S.; Yamasaki, K.; Komori, A.; Nagaoka, S.; Saeki, A.; Uchida, S.; Bekki, S.; Kugiyama, Y.; et al. Rapid Increase in Serum Low-Density Lipoprotein Cholesterol Concentration during Hepatitis C Interferon-Free Treatment. PLoS ONE 2016, 11, e0163644. [Google Scholar] [CrossRef]
- Zaid, A.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Chamberland, A.; Hamelin, J.; Tremblay, M.; Jacques, H.; Jin, W.; Davignon, J.; et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 2008, 48, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, J.L.; Gouni-Berthold, I.; Laufs, U. PCSK9 Inhibition: Insights From Clinical Trials and Future Prospects. Front. Physiol. 2020, 11, 595819. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Jankauskas, S.S.; Gambardella, J. Inclisiran: A new milestone on the PCSK9 road to tackle cardiovascular risk. Eur. Heart J. Cardiovasc. Pharm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef]
- Labonte, P.; Begley, S.; Guevin, C.; Asselin, M.C.; Nassoury, N.; Mayer, G.; Prat, A.; Seidah, N.G. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 2009, 50, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Bianconi, V.; Francisci, D.; Schiaroli, E.; Bagaglia, F.; Sahebkar, A.; Baldelli, F. Hepatitis C virus and proprotein convertase subtilisin/kexin type 9: A detrimental interaction to increase viral infectivity and disrupt lipid metabolism. J. Cell. Mol. Med. 2017, 21, 3150–3161. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, Q. Proprotein convertase subtilisin/kexin type 9 inhibits hepatitis C virus replication through interacting with NS5A. J. Gen. Virol. 2018, 99, 44–61. [Google Scholar] [CrossRef]
- Ramanathan, A.; Gusarova, V.; Stahl, N.; Gurnett-Bander, A.; Kyratsous, C.A. Alirocumab, a Therapeutic Human Antibody to PCSK9, Does Not Affect CD81 Levels or Hepatitis C Virus Entry and Replication into Hepatocytes. PLoS ONE 2016, 11, e0154498. [Google Scholar] [CrossRef] [Green Version]
- Fasolato, S.; Pigozzo, S.; Pontisso, P.; Angeli, P.; Ruscica, M.; Savarino, E.; De Martin, S.; Lupo, M.G.; Ferri, N. PCSK9 Levels Are Raised in Chronic HCV Patients with Hepatocellular Carcinoma. J. Clin. Med. 2020, 9, 3134. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Haberl, E.M.; Rein-Fischboeck, L.; Aslanidis, C. Adipokines in Liver Cirrhosis. Int. J. Mol. Sci. 2017, 18, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridge, S.H.; Sheridan, D.A.; Felmlee, D.J.; Crossey, M.M.; Fenwick, F.I.; Lanyon, C.V.; Dubuc, G.; Seidah, N.G.; Davignon, J.; Thomas, H.C.; et al. PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: Evidence for genotype-specific regulation of lipoprotein metabolism. J. Hepatol. 2015, 62, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Endo, D.; Satoh, K.; Shimada, N.; Hokari, A.; Aizawa, Y. Impact of interferon-free antivirus therapy on lipid profiles in patients with chronic hepatitis C genotype 1b. World J. Gastroenterol. 2017, 23, 2355–2364. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Eacho, P.I.; Knierman, M.D.; Troutt, J.S.; Konrad, R.J.; Yu, X.; Schroeder, K.M. Isolation and characterization of the circulating truncated form of PCSK9. J. Lipid Res. 2014, 55, 1505–1514. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Miyaaki, H.; Miuma, S.; Taura, N.; Motoyoshi, Y.; Akahoshi, H.; Nakamura, J.; Takahashi, Y.; Honda, T.; Yajima, H.; et al. Changes in serum LDL, PCSK9 and microRNA-122 in patients with chronic HCV infection receiving Daclatasvir/Asunaprevir. Biomed. Rep. 2019, 10, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiser, J.J.; Burton, J.R.; Anderson, P.L.; Everson, G.T. Review and management of drug interactions with boceprevir and telaprevir. Hepatology 2012, 55, 1620–1628. [Google Scholar] [CrossRef] [Green Version]
- Yen, Y.H.; Kuo, F.Y.; Chen, C.H.; Hu, T.H.; Lu, S.N.; Wang, J.H.; Hung, C.H. Ultrasound is highly specific in diagnosing compensated cirrhosis in chronic hepatitis C patients in real world clinical practice. Medicine 2019, 98, e16270. [Google Scholar] [CrossRef]
- Butt, A.A.; Xiaoqiang, W.; Budoff, M.; Leaf, D.; Kuller, L.H.; Justice, A.C. Hepatitis C virus infection and the risk of coronary disease. Clin. Infect. Dis. 2009, 49, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Liu, P.H.; Hsu, C.Y.; Hsia, C.Y.; Su, C.W.; He, Y.J.; Lee, Y.H.; Huang, Y.H.; Hou, M.C.; Huo, T.I. Current noninvasive liver reserve models do not predict histological fibrosis severity in hepatocellular carcinoma. Sci. Rep. 2018, 8, 15074. [Google Scholar] [CrossRef] [PubMed]
- Ragazzo, T.G.; Paranagua-Vezozzo, D.; Lima, F.R.; de Campos Mazo, D.F.; Pessoa, M.G.; Oliveira, C.P.; Alves, V.A.F.; Carrilho, F.J. Accuracy of transient elastography-FibroScan(R), acoustic radiation force impulse (ARFI) imaging, the enhanced liver fibrosis (ELF) test, APRI, and the FIB-4 index compared with liver biopsy in patients with chronic hepatitis C. Clinics 2017, 72, 516–525. [Google Scholar] [CrossRef]
- Rajan, M.R.; Sotak, M.; Barrenas, F.; Shen, T.; Borkowski, K.; Ashton, N.J.; Biorserud, C.; Lindahl, T.L.; Ramstrom, S.; Scholl, M.; et al. Comparative analysis of obesity-related cardiometabolic and renal biomarkers in human plasma and serum. Sci. Rep. 2019, 9, 15385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, P.; Ganz, P.; Ma, Y.; Scherzer, R.; Hur, S.; Weigel, B.; Grunfeld, C.; Deeks, S.; Wasserman, S.; Scott, R.; et al. HIV and Hepatitis C-Coinfected Patients Have Lower Low-Density Lipoprotein Cholesterol Despite Higher Proprotein Convertase Subtilisin Kexin 9 (PCSK9): An Apparent “PCSK9-Lipid Paradox”. J. Am. Heart Assoc. 2016, 5, e002683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, J.D.; Horton, J.D. Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9 and cholesterol biosynthesis in humans. J. Lipid Res. 2010, 51, 3359–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysa, J.A.; Ooi, T.C.; Proctor, S.D.; Vine, D.F. Nutritional and Lipid Modulation of PCSK9: Effects on Cardiometabolic Risk Factors. J. Nutr. 2017, 147, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Persson, L.; Cao, G.; Stahle, L.; Sjoberg, B.G.; Troutt, J.S.; Konrad, R.J.; Galman, C.; Wallen, H.; Eriksson, M.; Hafstrom, I.; et al. Circulating proprotein convertase subtilisin kexin type 9 has a diurnal rhythm synchronous with cholesterol synthesis and is reduced by fasting in humans. Arter. Thromb Vasc. Biol. 2010, 30, 2666–2672. [Google Scholar] [CrossRef] [Green Version]
- Nozue, T. Lipid Lowering Therapy and Circulating PCSK9 Concentration. J. Atheroscler. Thromb. 2017, 24, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Bucholz, E.M.; Rodday, A.M.; Kolor, K.; Khoury, M.J.; de Ferranti, S.D. Prevalence and Predictors of Cholesterol Screening, Awareness, and Statin Treatment Among US Adults with Familial Hypercholesterolemia or Other Forms of Severe Dyslipidemia (1999–2014). Circulation 2018, 137, 2218–2230. [Google Scholar] [CrossRef]
- Rao, G.A.; Pandya, P.K. Statin therapy improves sustained virologic response among diabetic patients with chronic hepatitis C. Gastroenterology 2011, 140, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, V.; Treuner-Kaueroff, T.; Seehofer, D.; Berg, T.; Becker, S.; Ceglarek, U.; Thiery, J.; Kaiser, T. Low PCSK9 levels are correlated with mortality in patients with end-stage liver disease. PLoS ONE 2017, 12, e0181540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Skill, N.; Marcus, V.; Deschenes, M.; Tan, X.; Bouteaud, J.; Negi, S.; Awan, Z.; Aikin, R.; Kwan, J.; et al. Decreased PCSK9 expression in human hepatocellular carcinoma. BMC Gastroenterol. 2015, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Li, S.; Xu, B.; Guo, H.; Zhang, S.; Cai, Y. Inhibition of Proprotein Convertase Subtilisin/Kexin Type 9 Ameliorates Liver Fibrosis via Mitigation of Intestinal Endotoxemia. Inflammation 2020, 43, 251–263. [Google Scholar] [CrossRef]
- Emma, M.R.; Giannitrapani, L.; Cabibi, D.; Porcasi, R.; Pantuso, G.; Augello, G.; Giglio, R.V.; Re, N.L.; Capitano, A.R.; Montalto, G.; et al. Hepatic and circulating levels of PCSK9 in morbidly obese patients: Relation with severity of liver steatosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158792. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Ferri, N.; Macchi, C.; Meroni, M.; Lanti, C.; Ricci, C.; Maggioni, M.; Fracanzani, A.L.; Badiali, S.; Fargion, S.; et al. Liver fat accumulation is associated with circulating PCSK9. Ann. Med. 2016, 48, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Bianconi, V.; Gragnano, F.; Moscarella, E.; Fimiani, F.; Monda, E.; Scudiero, O.; Limongelli, G.; Pirro, M.; Calabro, P. Beyond cholesterol metabolism: The pleiotropic effects of proprotein convertase subtilisin/kexin type 9 (PCSK9). Genetics, mutations, expression, and perspective for long-term inhibition. Biofactors 2020, 46, 367–380. [Google Scholar] [CrossRef]
- Toth, S.; Fedacko, J.; Pekarova, T.; Hertelyova, Z.; Katz, M.; Mughees, A.; Kuzma, J.; Stefanic, P.; Kopolovets, I.; Pella, D. Elevated Circulating PCSK9 Concentrations Predict Subclinical Atherosclerotic Changes in Low Risk Obese and Non-Obese Patients. Cardiol. Ther. 2017, 6, 281–289. [Google Scholar] [CrossRef]
- Giannelli, G.; Antonaci, S. Immunological and molecular aspects of liver fibrosis in chronic hepatitis C virus infection. Histol. Histopathol. 2005, 20, 939–944. [Google Scholar] [CrossRef]
- Babiker, A.; Hassan, M.; Muhammed, S.; Taylor, G.; Poonia, B.; Shah, A.; Bagchi, S. Inflammatory and cardiovascular diseases biomarkers in chronic hepatitis C virus infection: A review. Clin. Cardiol. 2020, 43, 222–234. [Google Scholar] [CrossRef]
- Ruscica, M.; Tokgozoglu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzl, M.W.; Resl, M.; Klammer, C.; Egger, M.; Dieplinger, B.; Clodi, M. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Is Not Induced in Artificial Human Inflammation and Is Not Correlated with Inflammatory Response. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyrina, A.; Olmstead, A.D.; Steven, P.; Krajden, M.; Tam, E.; Jean, F. Treatment-Induced Viral Cure of Hepatitis C Virus-Infected Patients Involves a Dynamic Interplay among three Important Molecular Players in Lipid Homeostasis: Circulating microRNA (miR)-24, miR-223, and Proprotein Convertase Subtilisin/Kexin Type 9. EBioMedicine 2017, 23, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, S.A.; Berge, K.E.; Leren, T.P. The unique role of proprotein convertase subtilisin/kexin 9 in cholesterol homeostasis. J. Intern. Med. 2009, 266, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.E.; Serper, M.A.; Mehta, R.; Fox, R.; John, B.; Aytaman, A.; Baytarian, M.; Hunt, K.; Albrecht, J.; Njei, B.; et al. Effects of Hypercholesterolemia and Statin Exposure on Survival in a Large National Cohort of Patients With Cirrhosis. Gastroenterology 2019, 156, 1693–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, M.H.; Tseng, C.W.; Lee, C.H.; Tseng, K.C. Drug-drug interactions between direct-acting antivirals and statins in the treatment of chronic hepatitis C. Tzu Chi Med. J. 2020, 32, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, J.; Niu, M.; Chen, X.; Gao, L.; Wu, Q.; Zheng, K.; Xu, K. Statins and the risk of cirrhosis in hepatitis B or C patients: A systematic review and dose-response meta-analysis of observational studies. Oncotarget 2017, 8, 59666–59676. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCV | Non-HCV | p-Value | |
|---|---|---|---|
| Number | 82 | 55 | ns |
| Sex (female/male) | 33/49 | 23/32 | ns |
| Age (years) | 59 (24–80) | 58 (21–80) | ns |
| BMI (kg/m2) | 27.1 (18.4–40.4) | 26.3 (17.8–39.7) | ns |
| MELD score | 8 (6–20) | na | |
| ALT (U/L) | 86 (22–287) | 32 (16–288) 54 | <0.001 |
| AST (U/L) | 75 (17–1230) | 24 (12–256) 54 | <0.001 |
| Albumin (g/L) | 36 (19–45) 81 | 47 (32–56) | <0.001 |
| Bilirubin (mg/dL) | 0.8 (0.3–4.3) | 0.5 (0.2–2.8) 53 | <0.001 |
| INR | 1.13 (0.91–2.44) | nd | |
| Creatinine (mg/dL) Ferritin (ng/mL) | 0.78 (0.14–1.31) 81 141 (7.0–2309) 76 | nd nd | |
| Leukocytes (n/L) | 5.9 (2.2–72.4) | nd | |
| CRP (mg/L) | 2.9 (2.9–72.4) | nd | |
| LDL (mg/dL) | 88 (31–210) 77 | 112 (44–340) | <0.01 |
| HDL (mg/dL) | 51 (19–103) 77 | nd | |
| Viral load (U/mL) | 1.4 × 106 | na | |
| Genotype 1a/1b/3a/rare | 24/38/14/6 | na | |
| FIB-4 (no fibrosis/intermediate values/yes) | 14/15/53 | na | |
| APRI (no fibrosis/no reliable values/fibrosis/cirrhosis) | 13/12/29/28 | na | |
| ARFI (F1/F3/F4) | 15/25/42 | na | |
| NAFLD (no fibrosis/no reliable values/fibrosis/nd) | 17/22/35/8 | na | |
| PCSK9 (ng/mL) | 218 (99–418) | 170 (40–360) | <0.01 |
| HCV Cirrhosis | Non-HCV Cirrhosis | |
|---|---|---|
| Number | 37 | 26 |
| Sex (female/male) | 15/22 | 5/21 |
| Age (years) | 61 (38–80) | 49 (40–81) |
| MELD score | 10 (7–20) | 8 (6–21) 22 |
| Albumin (g/L) | 34 (19–45) | 32 (2–4) |
| Bilirubin (mg/dL) | 1.2 (0.4–4.3) | 1.1 (0.4–3.7) |
| LDL (mg/dL) PCSK9 (ng/mL) | 68 (31–152) 171 (99–418) | 35 (17–81) 108 (41–290) *** |
| Parameter | Baseline (82 Patients) | 4-Weeks (79 Patients) | 12-Weeks (81 Patients) | 12 Weeks Posttreatment (76 Patients) |
|---|---|---|---|---|
| BMI, kg/m2 | 0.042 (0.720) | 0.037 (0.756) | 0.001 (0.995) | −0.026 (0.840) |
| Age | −0.248 (0.024) | −0.336 (0.002) | −0.180 (0.108) | −0.153 (0.221) |
| MELD score | −0.623 (<0.001) | −0.579 (<0.001) | −0.573 (<0.001) | −0.570 (<0.001) |
| ALT, U/L | −0.021 (0.853) | −0.042 (0.711) | −0.082 (0.467) | −0.060 (0.627) |
| AST, U/L | −0.260 (0.018) | −0.258 (0.022) | −0.288 (0.009) | −0.339 (<0.004) |
| Bilirubin, mg/dL | −0.506 (<0.001) | −0.587 (<0.001) | −0.597 (<0.001) | −0.568 (<0.001) |
| Albumin, g/L | 0.397 (<0.001) | 0.460 (<0.001) | 0.527 (<0.001) | 0.441 (<0.001) |
| INR | −0.595 (<0.001) | −0.549 (<0.001) | −0.608 (<0.001) | −0.574 (<0.001) |
| Creatinine, mg/dL | 0.074 (0.512) | −0.013 (0.909) | 0.010 (0.932) | 0.159 (0.191) |
| Leukocytes, n/L | 0.389 (<0.001) | 0.358 (0.001) | 0.503 (<0.001) | 0.383 (0.001) |
| CRP, mg/L HDL, mg/dL LDL, mg/dL | −0.288 (0.009) 0.047 (0.686) 0.502 (<0.001) | 0.218 (0.052) 0.042 (0.720) 0.513 (<0.001) | −0.235 (0.036) −0.048 (0.674) 0.578 (<0.001) | 0.018 (0.885) 0.022 (0.862) 0.595 (<0.001) |
| Viral load | 0.298 (0.007) | na | na | na |
| Parameter | Baseline (No Cirrhosis; 45 Patients) | Baseline (Cirrhosis; 37 Patients) | 12 Weeks Posttreatment (No Cirrhosis; 45 Patients) | 12 Weeks Posttreatment (Cirrhosis; 37 Patients) |
|---|---|---|---|---|
| Age | −0.010 (0.949) | −0.300 (0.071) | −0.168 (0.306) | −0.021 (0.913) |
| MELD score | −0.061 (0.689) | −0.707 (<0.001) | −0.195 (0.234) | −0.687 (<0.001) |
| ALT, U/L | −0.019 (0.900) | −0.332 (0.045) | −0.034 (0.836) | −0.009 (0.964) |
| AST, U/L | 0.063 (0.681) | −0.011 (0.951) | 0.047 (0.776) | −0.323 (0.082) |
| Bilirubin, mg/dL | −0.097 (0.525) | −0.653 (<0.001) | −0.294 (0.069) | −0.603 (0.001) |
| Albumin, g/L | −0.117 (0.451) | 0.442 (0.006) | −0.322 (0.049) | 0.283 (0.130) |
| INR | −0.083 (0.589) | −0.567 (<0.001) | −0.180 (0.274) | −0.567 (<0.001) |
| Leukocytes, n/L | −0.052 (0.735) | 0.495 (0.002) | −0.083 (0.615) | 0.367 (0.050) |
| CRP, mg/L LDL, mg/dL | −0.185 (0.223) 0.207 (0.193) | −0.526 (0.001) 0.316 (0.061) | −0.218 (0.182) 0.270 (0.123) | −0.526 (0.001) 0.459 (0.014) |
| Viral load | 0.326 (0.029) | −0.003 (0.987) | na | na |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grimm, J.; Peschel, G.; Müller, M.; Schacherer, D.; Wiest, R.; Weigand, K.; Buechler, C. Rapid Decline of Serum Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) in Non-Cirrhotic Patients with Chronic Hepatitis C Infection Receiving Direct-Acting Antiviral Therapy. J. Clin. Med. 2021, 10, 1621. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10081621
Grimm J, Peschel G, Müller M, Schacherer D, Wiest R, Weigand K, Buechler C. Rapid Decline of Serum Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) in Non-Cirrhotic Patients with Chronic Hepatitis C Infection Receiving Direct-Acting Antiviral Therapy. Journal of Clinical Medicine. 2021; 10(8):1621. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10081621
Chicago/Turabian StyleGrimm, Jonathan, Georg Peschel, Martina Müller, Doris Schacherer, Reiner Wiest, Kilian Weigand, and Christa Buechler. 2021. "Rapid Decline of Serum Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) in Non-Cirrhotic Patients with Chronic Hepatitis C Infection Receiving Direct-Acting Antiviral Therapy" Journal of Clinical Medicine 10, no. 8: 1621. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10081621