Systemic Redox Imbalance in Patients with Chronic Granulomatous Disease


 , , , and
, , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Population
2.2. Blood Samples
2.3. Redox Status
2.4. Redox Assays
2.5. Statistical Analysis
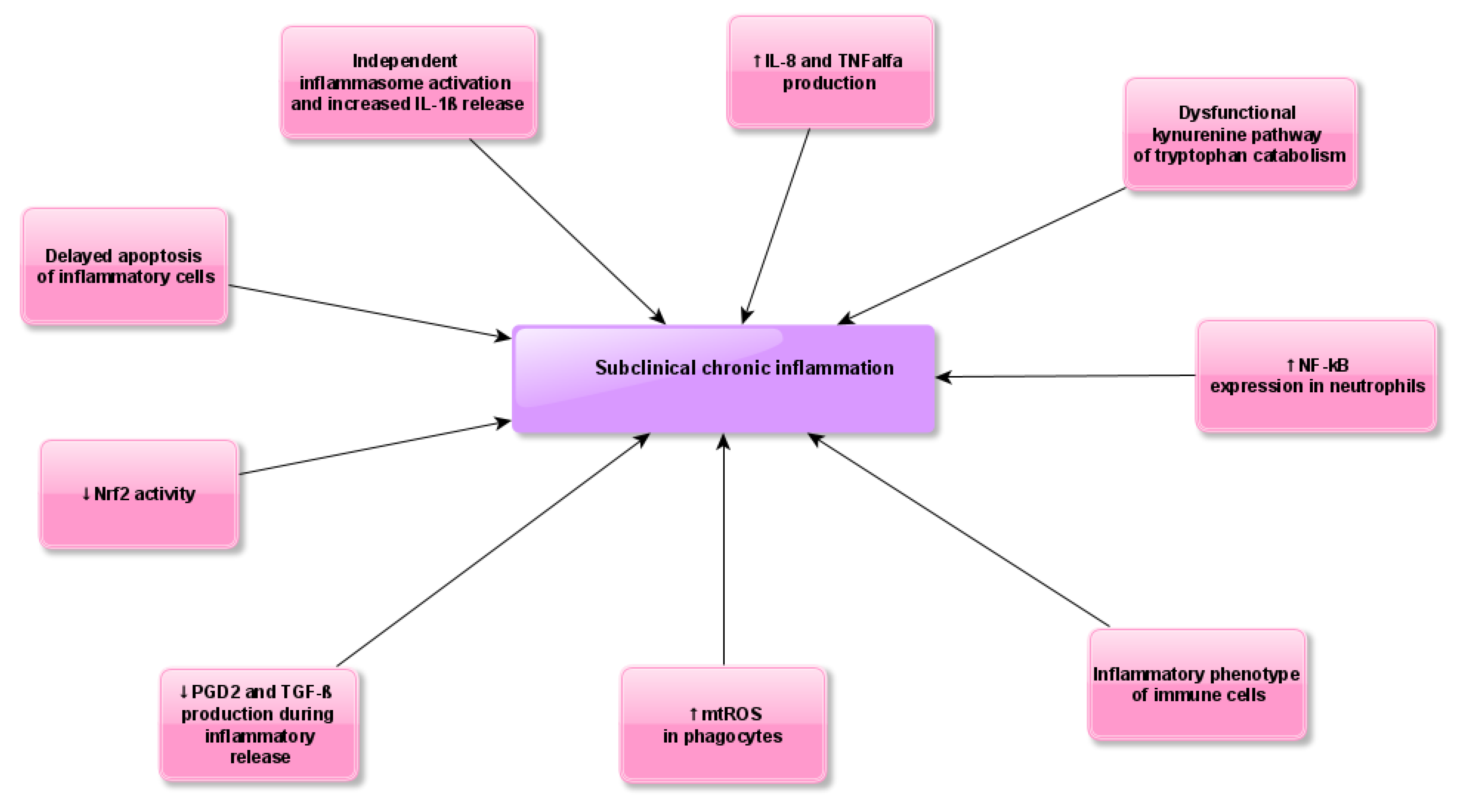
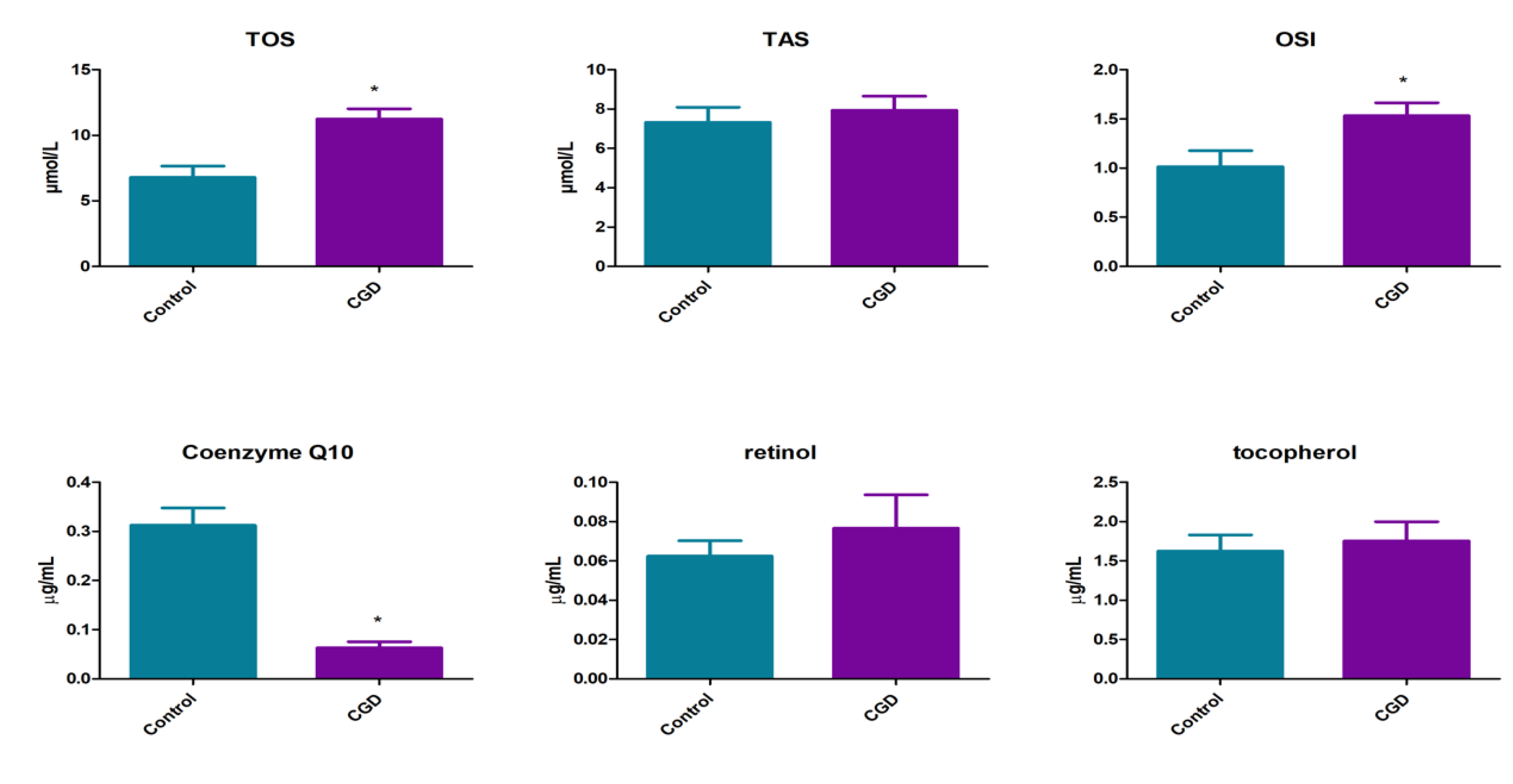
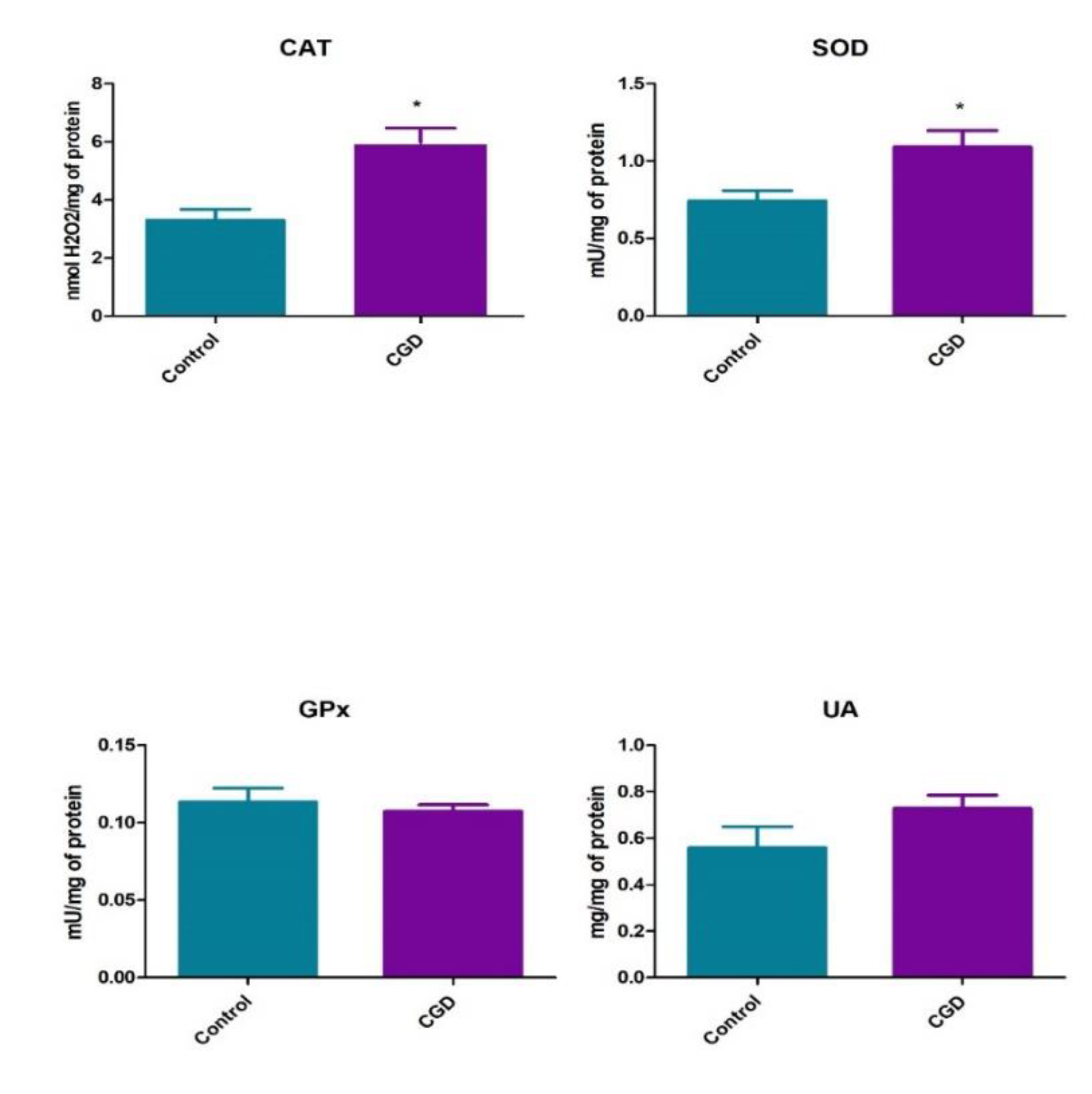
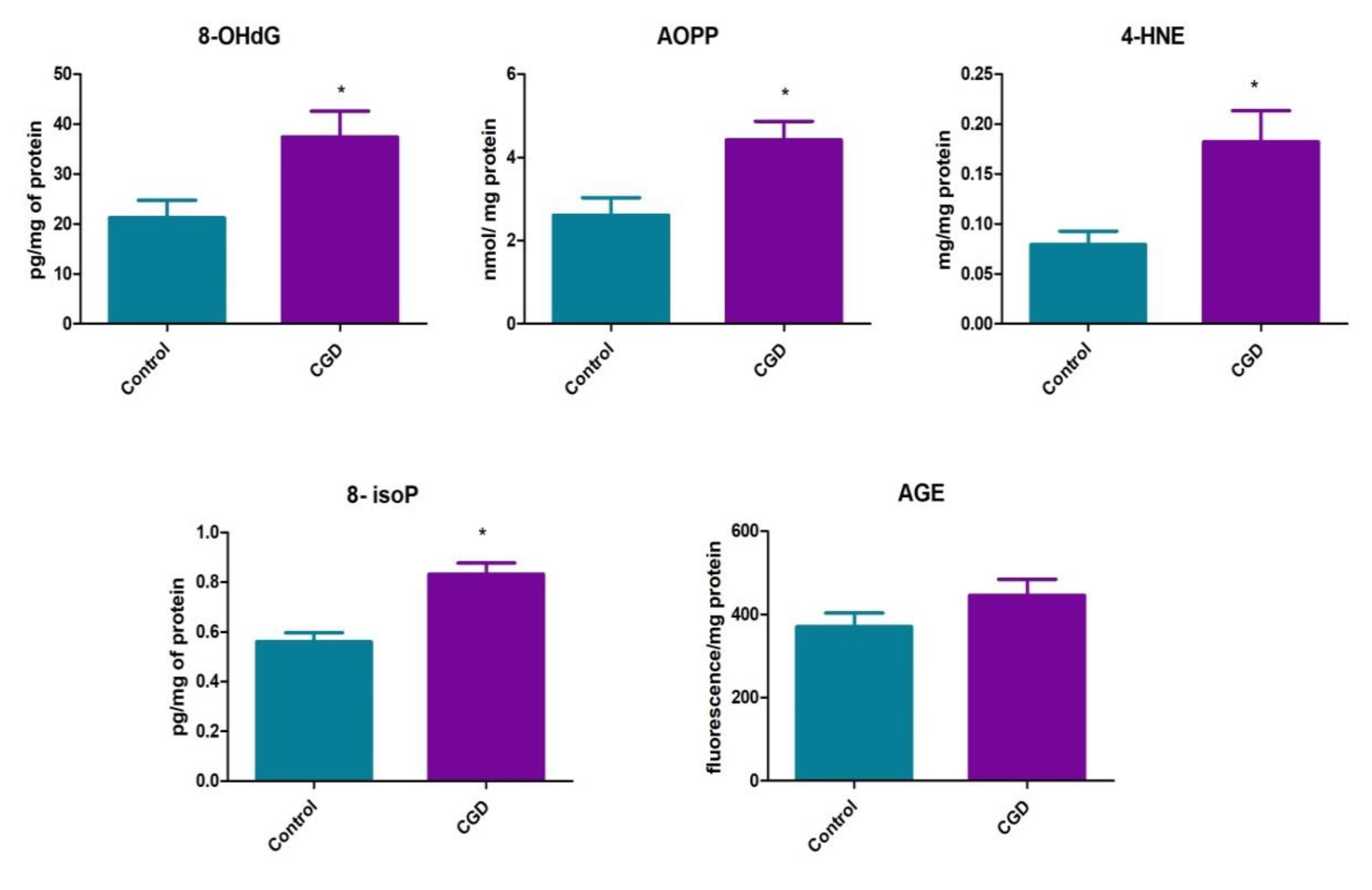
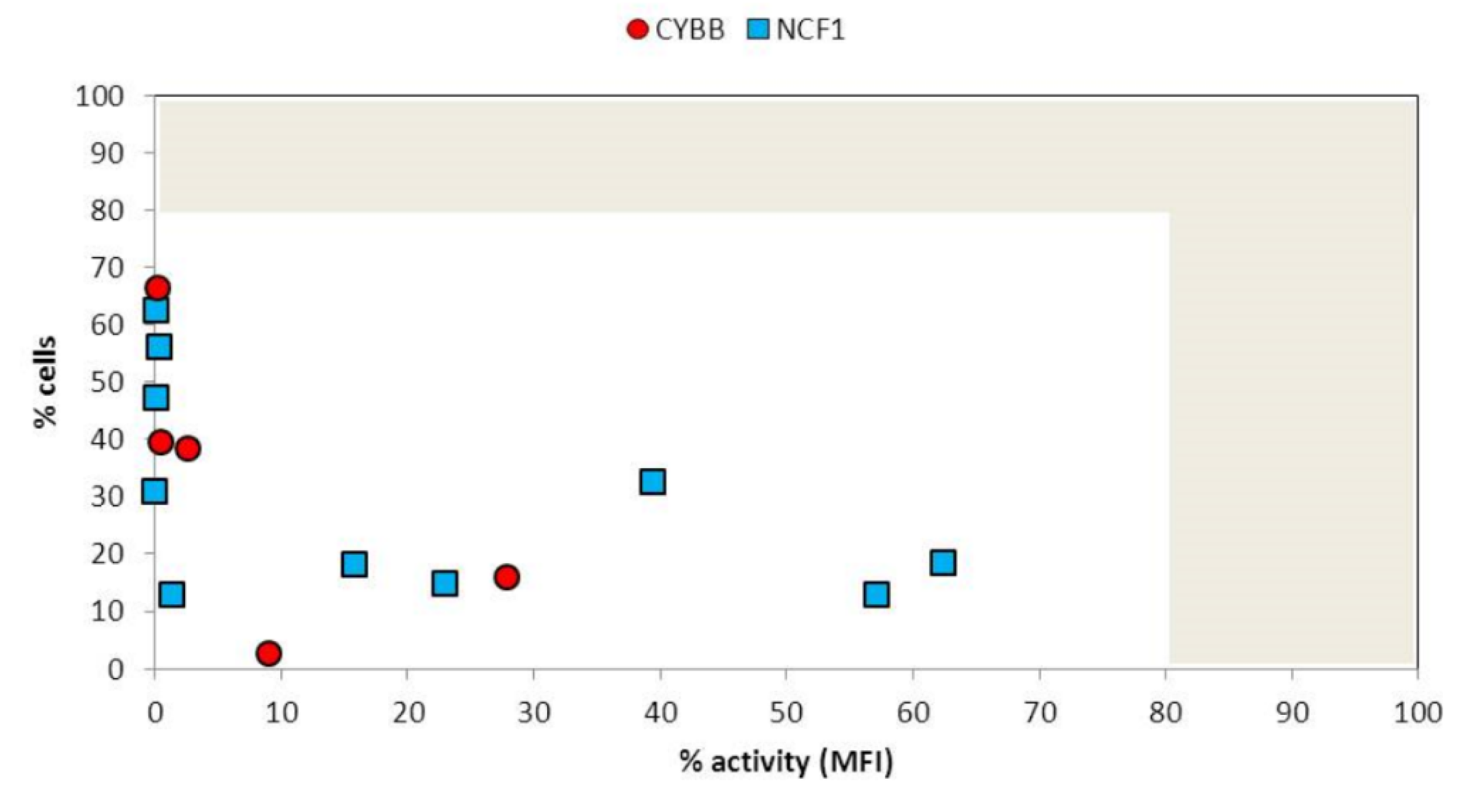
3. Results
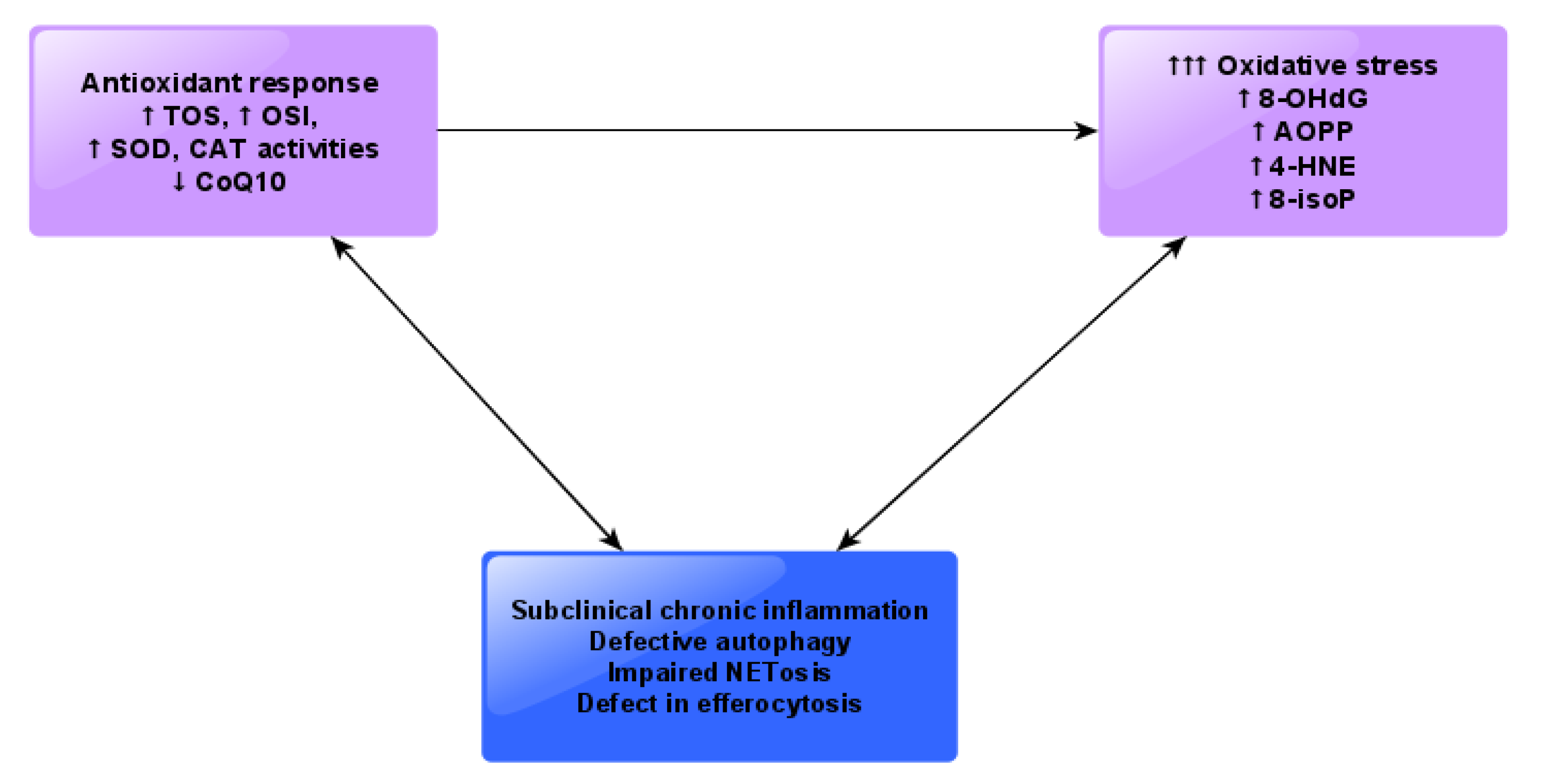
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltim.) 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Jongco, A.M.; Thomsen, K.F.; Garabedian, E.K.; Thomsen, I.P. Noninfectious manifestations and complications of chronic granulomatous disease. J. Pediatric Infect. Dis. Soc. 2018, 7, S18–S24. [Google Scholar] [CrossRef] [Green Version]
- Renzi, S.; Langenberg-Ververgaert, K.P.S.; Waespe, N.; Ali, S.; Bartram, J.; Michaeli, O.; Upton, J.; Cada, M. Primary immunodeficiencies and their associated risk of malignancies in children: An overview. Eur. J. Pediatr. 2020, 179, 689–697. [Google Scholar] [CrossRef]
- van der Weyden, L.; Speak, A.O.; Swiatkowska, A.; Clare, S.; Schejtman, A.; Santilli, G.; Arends, M.J.; Adams, D.J. Pulmonary metastatic colonisation and granulomas in NOX2-deficient mice. J. Pathol. 2018, 246, 300–310. [Google Scholar] [CrossRef]
- Thomas, D.C. How the phagocyte NADPH oxidase regulates innate immunity. Free Radic. Biol. Med. 2018, 125, 44–52. [Google Scholar] [CrossRef]
- Meissner, F.; Seger, R.A.; Moshous, D.; Fischer, A.; Reichenbach, J.; Zychlinsky, A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 2010, 116, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Sundqvist, M.; Christenson, K.; Björnsdottir, H.; Osla, V.; Karlsson, A.; Dahlgren, C.; Speert, D.P.; Fasth, A.; Brown, K.L.; Bylund, J. Elevated mitochondrial reactive oxygen species and cellular redox imbalance in human NADPH-oxidase-deficient phagocytes. Front. Immunol. 2017, 8, 1828. [Google Scholar] [CrossRef] [Green Version]
- Griffith, L.M.; Cowan, M.J.; Notarangelo, L.D.; Kohn, D.B.; Puck, J.M.; Shearer, W.T.; Burroughs, L.M.; Torgerson, T.R.; Decaluwe, H.; Haddad, E. Primary Immune Deficiency Treatment Consortium (PIDTC) update. J. Allergy Clin. Immunol. 2016, 138, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejczyk, M.; Zalewska, A.; Ładny, J.R. Salivary Antioxidant Barrier, Redox Status, and Oxidative Damage to Proteins and Lipids in Healthy Children, Adults, and the Elderly. Oxid. Med. Cell. Longev. 2019, 2019, 4393460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar] [PubMed]
- Aebi, H. [13] Catalase in Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar]
- Karpińska, J.; Mikołuć, B.; Motkowski, R.; Piotrowska-Jastrzebska, J. HPLC method for simultaneous determination of retinol, α-tocopherol and coenzyme Q10 in human plasma. J. Pharm. Biomed. Anal. 2006, 42, 232–236. [Google Scholar] [CrossRef]
- Kalousová, M.; Škrha, J.; Zima, T. Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiol. Res. 2002, 51, 597–604. [Google Scholar]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Romaniuk, W.; Markowski, A.; Kędra, B.; Zalewska, A.; Pryczynicz, A.; Matowicka-Karna, J.; Guzińska-Ustymowicz, K. Antioxidant Barrier, Redox Status, and Oxidative Damage to Biomolecules in Patients with Colorectal Cancer. Can Malondialdehyde and Catalase Be Markers of Colorectal Cancer Advancement? Biomolecules 2019, 9, 637. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Morel, A.; Saso, L.; Saluk, J. Isoprostanes and neuroprostanes as biomarkers of oxidative stress in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2014, 2014, 572491. [Google Scholar] [CrossRef]
- Janicka, M.; Kot-Wasik, A.; Kot, J.; Namieśnik, J. Isoprostanes-biomarkers of lipid peroxidation: Their utility in evaluating oxidative stress and analysis. Int. J. Mol. Sci. 2010, 11, 4631–4659. [Google Scholar] [CrossRef] [Green Version]
- Basu, S. Bioactive eicosanoids: Role of prostaglandin F2α and F2-isoprostanes in inflammation and oxidative stress related pathology. Mol. Cells 2010, 30, 383–391. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Zalewska, A.; Chabowski, A. Redox balance, antioxidant defense, and oxidative damage in the hypothalamus and cerebral cortex of rats with high fat diet-induced insulin resistance. Oxid. Med. Cell. Longev. 2018, 2018, 6940515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.J.; Bittelmeyer, A.M.; Raess, B.U. Irreversible inhibition of plasma membrane (Ca2+ + Mg2+)-ATPase and Ca2+ transport by 4-OH-2,3-trans-nonenal. Arch. Biochem. Biophys. 1999, 361, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Sanguigni, V.; Carnevale, R.; Plebani, A.; Rossi, P.; Finocchi, A.; Pignata, C.; De Mattia, D.; Martire, B.; Pietrogrande, M.C.; et al. Hereditary deficiency of gp91phox is associated with enhanced arterial dilatation: Results of a multicenter study. Circulation 2009, 120, 1616–1622. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, R.; Loffredo, L.; Sanguigni, V.; Plebani, A.; Rossi, P.; Pignata, C.; Martire, B.; Finocchi, A.; Pietrogrande, M.C.; Azzari, C.; et al. Different degrees of NADPH oxidase 2 regulation and in vivo platelet Activation: Lesson from chronic granulomatous disease. J. Am. Heart Assoc. 2014, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin resistance and oxidative stress in the brain: What’s new? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.J.; Niu, H.X.; Hou, F.F.; Zhang, L.; Fu, N.; Nagai, R.; Lu, X.; Chen, B.H.; Shan, Y.X.; Tian, J.W.; et al. Advanced oxidation protein products activate vascular endothelial cells via a RAGE-mediated signaling pathway. Antioxid. Redox Signal. 2008, 10, 1699–1712. [Google Scholar] [CrossRef]
- Zalewska, A.; Maciejczyk, M.; Szulimowska, J.; Imierska, M.; Błachnio-Zabielska, A. High-fat diet affects ceramide content, disturbs mitochondrial redox balance, and induces apoptosis in the submandibular glands of mice. Biomolecules 2019, 9, 877. [Google Scholar] [CrossRef] [Green Version]
- Scavuzzi, B.M.; Simão, A.N.C.; Iriyoda, T.M.V.; Lozovoy, M.A.B.; Stadtlober, N.P.; Franchi Santos, L.F.D.R.; Flauzino, T.; de Medeiros, F.A.; de Sá, M.C.; Consentin, L.; et al. Increased lipid and protein oxidation and lowered anti-oxidant defenses in systemic lupus erythematosus are associated with severity of illness, autoimmunity, increased adhesion molecules, and Th1 and Th17 immune shift. Immunol. Res. 2018, 66, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardino, G.; Cicalese, M.P.; Delmonte, O.; Migliavacca, M.; Palterer, B.; Loffredo, L.; Cirillo, E.; Gallo, V.; Violi, F.; Pignata, C. NADPH oxidase deficiency: A multisystem approach. Oxid. Med. Cell. Longev. 2017, 2017, 4590127. [Google Scholar] [CrossRef]
- Gabrion, A.; Hmitou, I.; Moshous, D.; Neven, B.; Lefèvre-Utile, A.; Diana, J.S.; Suarez, F.; Picard, C.; Blanche, S.; Fischer, A.; et al. Mammalian target of rapamycin inhibition counterbalances the inflammatory status of immune cells in patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 2017, 139, 1641–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejczyk, M.; Heropolitanska-Pliszka, E.; Pietrucha, B.; Sawicka-Powierza, J.; Bernatowska, E.; Wolska-Kusnierz, B.; Pac, M.; Car, H.; Zalewska, A.; Mikoluc, B. Antioxidant defense, redox homeostasis, and oxidative damage in children with ataxia telangiectasia and nijmegen breakage syndrome. Front. Immunol. 2019, 10, 2322. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Zalewska, A.; Ziembicka, D.; Zendzian-Piotrowska, M.; MacIejczyk, M. The impact of high-fat diet on mitochondrial function, free radical production, and nitrosative stress in the salivary glands of wistar rats. Oxid. Med. Cell. Longev. 2019, 2019, 2606120. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic. Res. 2019, 53, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Schlotte, V.; Sevanian, A.; Hochstein, P.; Weithmann, K.U. Effect of uric acid and chemical analogues on oxidation of human low density lipoprotein in vitro. Free Radic. Biol. Med. 1998, 25, 839–847. [Google Scholar] [CrossRef]
- Pasalic, D.; Marinkovic, N.; Feher-turkovic, L. Uric acid as one of the important factors in multifactorial disorders—Facts and controversies. Biochem. Med. 2012, 22, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Raizner, A.E. Coenzyme Q10. Methodist Debakey Cardiovasc. J. 2019, 15, 185. [Google Scholar]
- López, L.C.; Luna-Sánchez, M.; García-Corzo, L.; Quinzii, C.M.; Hirano, M. Pathomechanisms in coenzyme Q10-deficient human fibroblasts. Mol. Syndromol. 2014, 5, 163–169. [Google Scholar] [CrossRef]
- Rodick, T.C.; Seibels, D.R.; Babu, J.R.; Huggins, K.W.; Ren, G.; Mathews, S.T. Potential role of coenzyme Q10 in health and disease conditions. Nutr. Diet. Suppl. 2018, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Montero, R.; Yubero, D.; Salgado, M.C.; González, M.J.; Campistol, J.; O’Callaghan, M.D.M.; Pineda, M.; Delgadillo, V.; Maynou, J.; Fernandez, G.; et al. Plasma coenzyme Q10 status is impaired in selected genetic conditions. Sci. Rep. 2019, 9, 793. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, M.-J.; Lee, S.; Cho, M.-L. Coenzyme Q10 Exerts Anti-Inflammatory Activity and Induces Treg in Graft versus Host Disease. J. Med. Food 2016, 19, 238–244. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Lee, S.H.; Yang, E.-J.; Kim, J.-K.; Kim, E.-K.; Jung, K.; Jung, H.; Lee, K.; Lee, H.H.; Lee, B.-I.; et al. Coenzyme Q10 Inhibits Th17 and STAT3 Signaling Pathways to Ameliorate Colitis in Mice. J. Med. Food 2017, 20, 821–829. [Google Scholar] [CrossRef]
- Alcocer-Gómez, E.; Culic, O.; Navarro-Pando, J.M.; Sánchez-Alcázar, J.A.; Bullón, P. Effect of Coenzyme Q10 on Psychopathological Symptoms in Fibromyalgia Patients. CNS Neurosci. Ther. 2017, 23, 188–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Precursor Ion (m/z) | Product Ion (m/z) | Collision Energy [eV] |
|---|---|---|---|
| Retinol | 269.10 | 213.20 93.10 | −12 −23 |
| α-Tocopherol | 429.30 | 165.10 137.05 | −25 −48 |
| Coenzyme Q10 | 863.60 | 197.15 109.10 | −45 −47 |
| No. | Sex (Male/Female) | Age, Years | Genetic Mutation | Age at CDG Diagnosis, Years | Clinical Manifestations Leading to Diagnosis of CGD |
|---|---|---|---|---|---|
| 1 | M | 25 | CYBB | 6 | Skin abscess |
| 2 | M | 25 | CYBB | 5 | Brain abscess; liver abscess; rectal abscess; pneumonitis; myocarditis; erythema nodosum |
| 3 | M | 4 | CYBB | 2 | Perianal abscesses |
| 4 | M | 26 | CYBB | 1 | Lymph nodes and skin abscesses; several with pneumonitis |
| 5 | M | 28 | CYBB | 10 | Pneumonia |
| 6 | M | 39 | NCF1 | 13 | Liver abscess |
| 7 | F | 24 | NCF1 | 17 | Liver abscesses; perianal abscesses |
| 8 | F | 9 | NCF1 | 7 | Lymphadenitis; |
| 9 | F | 13 | NCF1 | 11 | Recurrent pneumonia |
| 10 | F | 6 | NCF1 | 5 | Lymphadenitis; rectal abscess |
| 11 | M | 2 | NCF1 | 1 | Pneumonia; pulmonary aspergillosis |
| 12 | M | 36 | NCF1 | 34 | Syphilis; skin abscesses; staphylococcal abscess of the brain and lungs |
| 13 | F | 12 | NCF1 | 10 | Mulch pneumonia |
| 14 | M | 24 | NCF1 | 20 | Liver abscess |
| 15 | M | 26 | NCF1 | 6 | Liver abscess; tuberculosis of the lymph nodes |
| Parameter | CYBB, n = 5 | NCF1, n = 10 | p-Values |
|---|---|---|---|
| TAS, µmol/L | 6.6 ± 1.8 | 8.5 ± 3.1 | ns |
| TOS, µmol/L | 10.6 ± 4.1 | 11.6 ± 2.3 | ns |
| OSI | 1.6 ± 0.7 | 1.5 ± 0.4 | ns |
| CAT, nmol/100 mg of protein | 6.2 ± 1.9 | 5.8 ± 1.9 | ns |
| SOD, mU/mg | 1.1 ± 0.2 | 1.1 ± 0.5 | ns |
| GP, mU/100 mg protein | 0.1± 0.01 | 0.1 ± 0,02 | ns |
| UA, µg/100 mg of protein | 0.8 ± 0.2 | 0.7 ± 0.2 | ns |
| Coenzyme Q10, μg/mL | 0.08 ± 0.04 | 0.05 ± 0.05 | ns |
| Retinol, μg/mL | 0.1 ± 0.09 | 0.08 ± 0.07 | ns |
| Tocopherol, μg/mL | 1.9 ± 1.1 | 1.6 ± 0.86 | ns |
| 8-OHdG, pg/mg of protein | 18.0 ± 28.7 | 24.7 ± 19.8 | ns |
| AOPP, nmol/mg protein | 4.1 ± 1.5 | 4.5 ± 2 | ns |
| 4-HNE, µg/mg protein | 0.17 ± 0.14 | 0.19 ± 0.06 | ns |
| 8-isoP, pg/mg protein | 0.9 ± 0.2 | 0.8 ± 0.2 | ns |
| AGE, fluorescence/mg protein | 500.2 ± 168.8 | 443 ± 153 | ns |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heropolitanska-Pliszka, E.; Berk, K.; Maciejczyk, M.; Sawicka-Powierza, J.; Bernatowska, E.; Wolska-Kusnierz, B.; Pac, M.; Dabrowska-Leonik, N.; Piatosa, B.; Lewandowicz-Uszynska, A.; et al. Systemic Redox Imbalance in Patients with Chronic Granulomatous Disease. J. Clin. Med. 2020, 9, 1397. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051397
Heropolitanska-Pliszka E, Berk K, Maciejczyk M, Sawicka-Powierza J, Bernatowska E, Wolska-Kusnierz B, Pac M, Dabrowska-Leonik N, Piatosa B, Lewandowicz-Uszynska A, et al. Systemic Redox Imbalance in Patients with Chronic Granulomatous Disease. Journal of Clinical Medicine. 2020; 9(5):1397. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051397
Chicago/Turabian StyleHeropolitanska-Pliszka, Edyta, Klaudia Berk, Mateusz Maciejczyk, Jolanta Sawicka-Powierza, Ewa Bernatowska, Beata Wolska-Kusnierz, Malgorzata Pac, Nel Dabrowska-Leonik, Barbara Piatosa, Aleksandra Lewandowicz-Uszynska, and et al. 2020. "Systemic Redox Imbalance in Patients with Chronic Granulomatous Disease" Journal of Clinical Medicine 9, no. 5: 1397. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051397