
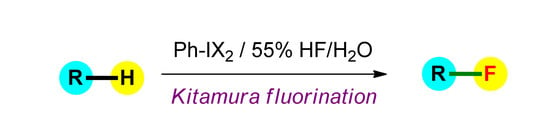
Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine
by
 , , and
, , and
Jianlin Han
1,* ,
,
Greg Butler
2,
Hiroki Moriwaki
3,
Hiroyuki Konno
4,
Vadim A. Soloshonok
5,6,* and
Tsugio Kitamura
7 1
Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, Jiangsu, China
2
Oakwood Chemical, Inc. 730 Columbia Hwy. N, Estill, SC 29918, USA
3
Hamari Chemical Ltd., 1-4-29 Kunijima, Higashi-Yodogawa-ku, Osaka 533-0024, Japan
4
Department of Biological Engineering, Graduate School of Science and Engineering, Yamagata University, Yonezawa, Yamagata 992-8510, Japan
5
Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, 20018 San Sebastián, Spain
6
IKERBASQUE, Basque Foundation for Science, Plaza Bizkaia, 48011 Bilbao, Spain
7
Department of Chemistry and Applied Chemistry, Saga University, 1 Honjo-machi, Saga 840-8502, Japan
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(9), 2116; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092116
Submission received: 4 April 2020
/
Revised: 22 April 2020
/
Accepted: 27 April 2020
/
Published: 30 April 2020
(This article belongs to the Special Issue Exclusive Papers of the Editorial Board Members of the Organic Chemistry Section of Molecules)
Abstract
:This review article focused on the innovative procedure for electrophilic fluorination using HF and in situ generation of the required electrophilic species derived from hypervalent iodine compounds. The areas of synthetic application of this approach include fluorination of 1,3-dicarbonyl compounds, aryl-alkyl ketones, styrene derivatives, α,β-unsaturated ketones and alcohols, homoallyl amine and homoallyl alcohol derivatives, 3-butenoic acids and alkynes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Over the last two decades, the chemistry of fluorine-containing compounds has emerged as one of the exciting areas of multidisciplinary research. The most notable impact of fluorine can be seen in materials [1,2,3,4,5,6,7,8,9,10], agriculture [11,12,13] and health-related industries [14,15,16,17,18,19,20,21,22]. To sustain the continuous advancement and pace of the innovations enabled by fluorine, many research groups are focusing on new methodological inventions allowing for more selective and economical syntheses of structurally diverse fluoro-organic compounds [23,24,25,26,27,28]. For instance, the recent progress in asymmetric synthesis of fluorine-containing tailor-made amino acids [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] was stimulated by growth in their applications to drug design [49] magnetic resonance imaging [50,51], positron emission tomography [52,53], and peptide/protein engineering [54,55,56,57,58,59,60]. Nevertheless, while some complex polyfunctional fluorine-containing molecules possessing useful properties represent the ultimate target of synthetic chemistry, more fundamental research still focuses on the formation of the C–F bond. In this regard, electrophilic fluorination is one of the most pioneering and rapidly developing areas of study [61,62,63,64,65,66,67]. Conceptually, this approach requires a carbon-centered nucleophile and an electrophilic source of fluorine. While the former is a well-established chemical unit, the “electrophilic fluorine” is still a rather exotic and mechanistically controversial entity [68,69,70,71]. The major thrust of research activity in this field was centered on the development of the corresponding reagents capable of releasing the required “electrophilic fluorine”. The most successful results have been achieved utilizing compounds with N–F bonds. For example, as presented in Figure 1, 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (1), introduced by Professor E. Banks [72], also known as Selectfluor, is one of the best reagents used as a source of “electrophilic fluorine”. Other reagents, developed by Professor N. Shibata [73,74,75,76], are N-fluoro-N-(methylsulfonyl)methanesulfonamide (Me-NFSI) (2), 3,5-di-tert-butyl-N-((3,5-di-tert-butyl-4-methoxyphenyl)sulfonyl)-N-fluoro-4-methoxybenzenesulfonamide (NFBSI) (3) and axially chiral NFSIs (4), which can be used for enantioselective fluorination [77]. All these reagents are shelf-stable, easy to handle and operationally convenient to use for various synthetic applications [78,79,80,81]. However, the preparation of N-F reagents usually requires molecular fluorine [72,82], rendering them rather expensive and not practical for large-scale syntheses.
From the standpoint of practicality, the application of HF, as a source of fluorine, with the in situ formation of the electrophilic species, would offer an attractive alternative as a general methodology for “electrophilic” formation of a C–F bond. In this review article, we discuss the use of hypervalent iodine compounds, as electrophilic centers, and HF as a source of fluorine, creatively assembled in a one-pot sequence, allowing one to perform “electrophilic fluorination” of various types of organic compounds. The synthetic generality and practicality of these methods is critically discussed.
2. Fluorination of 1,3-Dicarbonyl Compounds
2.1. Iodosylbenzene-Mediated Fluorination
In 2011, Professor T. Kitamura’s group has reported that mixing ethyl 3-oxo-3-phenylpropionate (5) with 1.2 equivalents of a hypervalent iodine compound and excess (10-fold) of 55% aqueous HF resulted in formation of the corresponding ethyl 2-fluoro-3-oxo-3-phenylpropionate (6) in up to 98% chemical yield (Scheme 1) [83].
It was found that the nature of a hypervalent iodine compound played a key role in the reaction, affording an excellent yield of 98% of product 6 with the application of iodosylbenzene (PhIO). The outcome of this reaction, the substitution of acidic hydrogen in 5 by fluorine, is classified as “electrophilic” fluorination and was previously reported using F2 [84], XeF2 [85,86,87] fluoroxy compounds [88,89,90,91,92,93,94], N–F compounds [95,96,97,98,99,100,101,102] of type 1–4 (Figure 1) and (difluoroiodo)toluene [103,104]; all of them are typical electrophilic fluorination reagents.
The mechanistic rationale for the reaction is presented in Scheme 2. It postulates the in situ formation of (difluoroiodo)benzene 7 from iodosylbenzene and two equivalents of hydrogen fluoride. The reaction of 7 with the enol 8 gives rise to intermediate 9, followed by the nucleophilic substitution of the iodine species to afford fluorinated compound 6 along with iodobenzene 10 as the final reaction products.
Some support for the proposed mechanistic pathway can be derived from the reactions of iodonium ylides with HF and HCl [105]. As shown in Scheme 3, iodonium ylide 11 can be prepared in quantitative yield by the reaction of keto ester 5 with (diacetoxyiodo)benzene in the presence of KOH in MeCN [106].
The reaction of iodonium ylide 11 with concentrated aqueous HCl, conducted in dichloromethane at ambient temperature, gave chlorinated product 12 in 55% yield [105]. Similar yields of fluorination product 6 were also obtained under the same conditions in the reactions of 11 with various HF reagents, such as 55% aqueous HF and TEA·3HF [105]. The noticeably lower chemical yields of 6 obtained in the reactions of iodonium ylide 11 with HF reagents (vs 98%), as compared with the reactions of 5 with HF in the presence of PhIO (Scheme 1 and Scheme 2) can be explained by enolization of intermediate 13 to vinyliodonium salt 14, further reactions of which give complex mixtures of products.
Meticulous investigations of HF/PhIO reactivity revealed that this new approach has a general synthetic application for fluorination of various compounds bearing the acidic CH2 moiety, such as 3-keto-esters, 1,3-diketones and malonic acid derivatives, summarized in Scheme 4.
Optimization of the reaction conditions and reagents revealed that besides the original iodosylbenzene, its derivatives, such as p- and o-iodosyltoluene, can also be used successfully, and in some cases, even give better chemical yields as compared with unsubstituted iodosylbenzene [107,108]. For example, fluorination of amide derivatives of 3-keto-esters 15 (R’ = CONAlk2) with HF/PhIO affords the corresponding products 16 with relatively low (<50%) yield, while the application of o-iodosyltoluene allows for improved yields of the target products 16 (up to 93%). The source of the HF has also been examined in detail showing that other HF reagents such as complexes with triethylamine and pyridine can be successfully used in place of aqueous HF. Of particular importance is the wide synthetic generality of this approach. Thus, 3-keto esters 15 bearing an aromatic ring with electron-withdrawing or donating substituents can be successfully used as substrates. In the case of derivatives 15 featuring alkyl groups (R and/or R’ = Alk) the chemical yields of the corresponding fluorinated products 16 are a bit lower (~50–70%), likely due to possible enolization.
2.2. Catalytic Iodoarene-Mediated Fluorination
Considering the postulated reaction mechanism (Scheme 2) involving eventual transformation (reduction) of iodosylbenzene to iodobenzene 10, the final reaction product, the authors posited that providing in situ efficient oxidation of the latter to iodosylbenzene, would allow for a catalytic version of this process. Indeed, a significant breakthrough was made with the application of m-CPBA as a terminal oxidant [109]. As presented in Scheme 5, the Ar-I, used in catalytic amounts, undergoes a three-step transformation: oxidation to Ar-IO, reaction with HF to produce Ar-IF2 and reaction with the enolate form of 15 giving rise to the target product 16.
The optimized conditions for this catalytic “electrophilic” fluorination method are presented in Scheme 6 and included the following: as low as 20 mol% ArI, 55% aq. HF as a source of fluorine, m-CPBA as the oxidizing reagent and 1,2-dichloroethane as a solvent. The reactions are conducted at 49 °C and can be easily scaled up. The overall synthetic generality of this catalytic process is rather similar to the stoichiometric version.
The catalytic version can be used for fluorination of 3-keto-esters and 1,3-diketones bearing substituted aromatic, aliphatic, ester as well as amide groups. It should be notated, however, that the yields of target products 16 are a bit lower as compared with the results obtained in the reactions using a stoichiometric amount of the corresponding iodoarene. Furthermore, in most of the cases, application of o-Tol-I gave the best conversion of the starting 1,3-dicarbonyl compounds 15 and highest yields of fluorinated products 16.
3. Fluorination of Aryl-Alkyl Ketones
Synthesis of fluorinated derivatives of monocarbonyl compounds, such as ketones and aldehydes, are of high importance in fluoro-organic chemistry [110,111,112,113,114,115,116,117,118,119,120,121]. These types of fluorinated derivatives are of proven synthetic value as building blocks for the preparation of a variety of polyfunctional fluorine-containing compounds of biological interest [30,122,123,124,125,126,127,128]. In particular, these derivatives can be easily transformed to the corresponding fluorinated amines and amino acids via biomimetic transamination [129,130,131]. General approaches for the preparation of α-fluoro-ketones include the following: two-step sequence of halogenation followed by nucleophilic substitution using fluoride [132,133,134,135,136] or a two-step process, involving the generation of the corresponding enolates, followed by fluorination using typical electrophilic fluorination reagents [137,138,139].
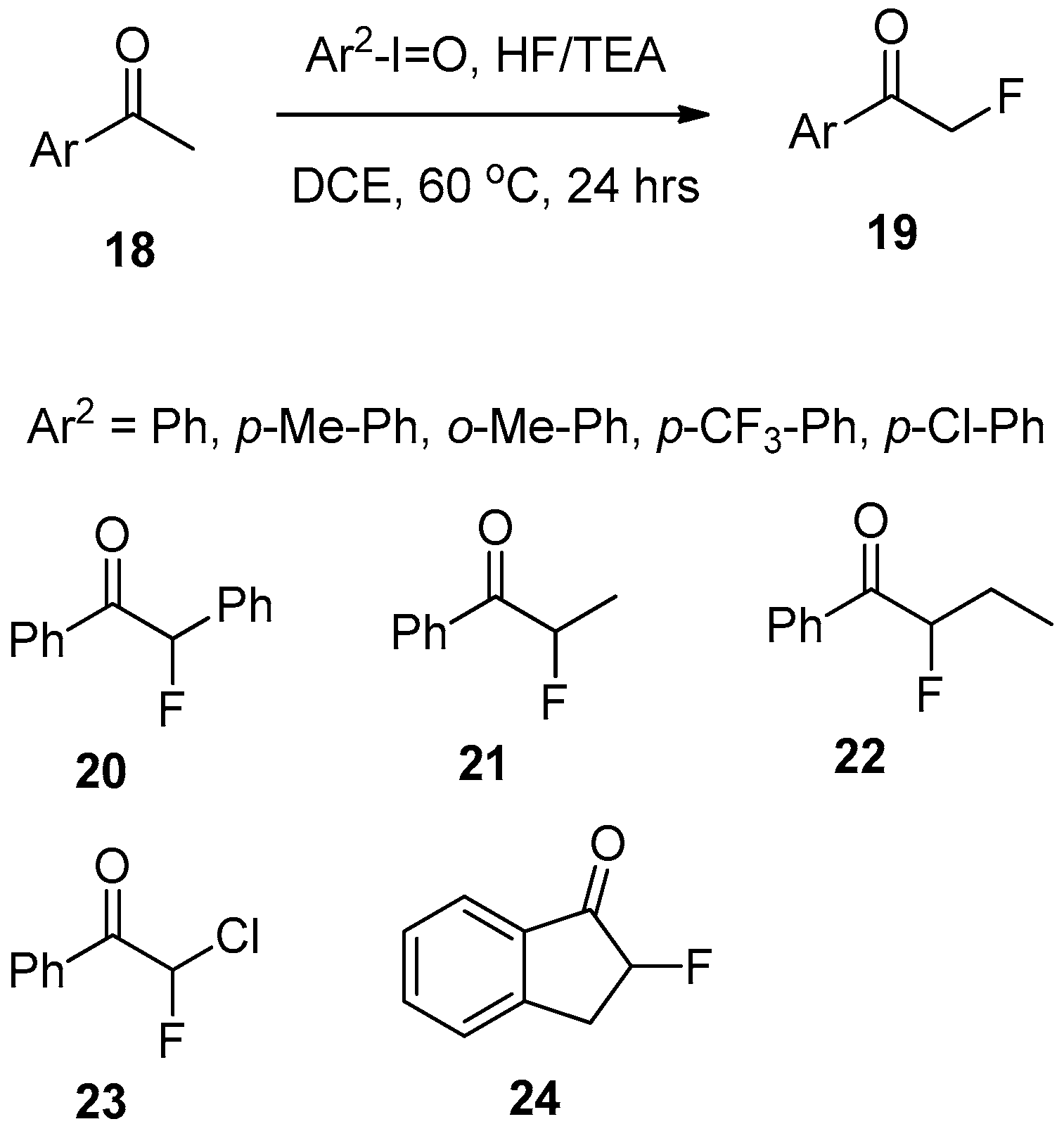
It was found that the direct application of Kitamura’s approach, using PhIO/hydrofluoric acid, for fluorination of acetophenone (Scheme 7) gives very low yields of the target product 17.
However, careful examination of various reaction conditions and reagents allowed for the discovery that the presence of water is detrimental for the fluorination process. The most likely reason is a reduction of fluoride’s nucleophilicity by coordination with water molecules. Consequently, it was found that the application of a triethylamine/5HF complex as a fluorine source provides for a smooth fluorination process affording α-fluoro-acetophenone 17 in a good yield [140]. Further research revealed that this reaction can be generally applied for various aryl-alkyl ketones (Scheme 8). Among possible sources of hypervalent iodine, p-iodosyltoluene (4-MeC6H4IO), p-chloloiodosylbenzene (4-ClC6H4IO), and p-iodosyl(trifluoromethyl)benzene (4-CF3C6H4IO) gave generally good results. Optimized reaction conditions included TEA/5HF in DCE (1,2-dichloroethane) at 60 °C for 24 h. Under these conditions, chemical yields of aryl-fluoromethyl ketones 19 varied between 70–85%. Of particular interest is the application of this reaction for α-fluorination of substrates derived from aryl-benzyl ketone 20, higher alkyl derivatives such as 21 and 22, haloalkyl 23 and cyclic compounds 24.
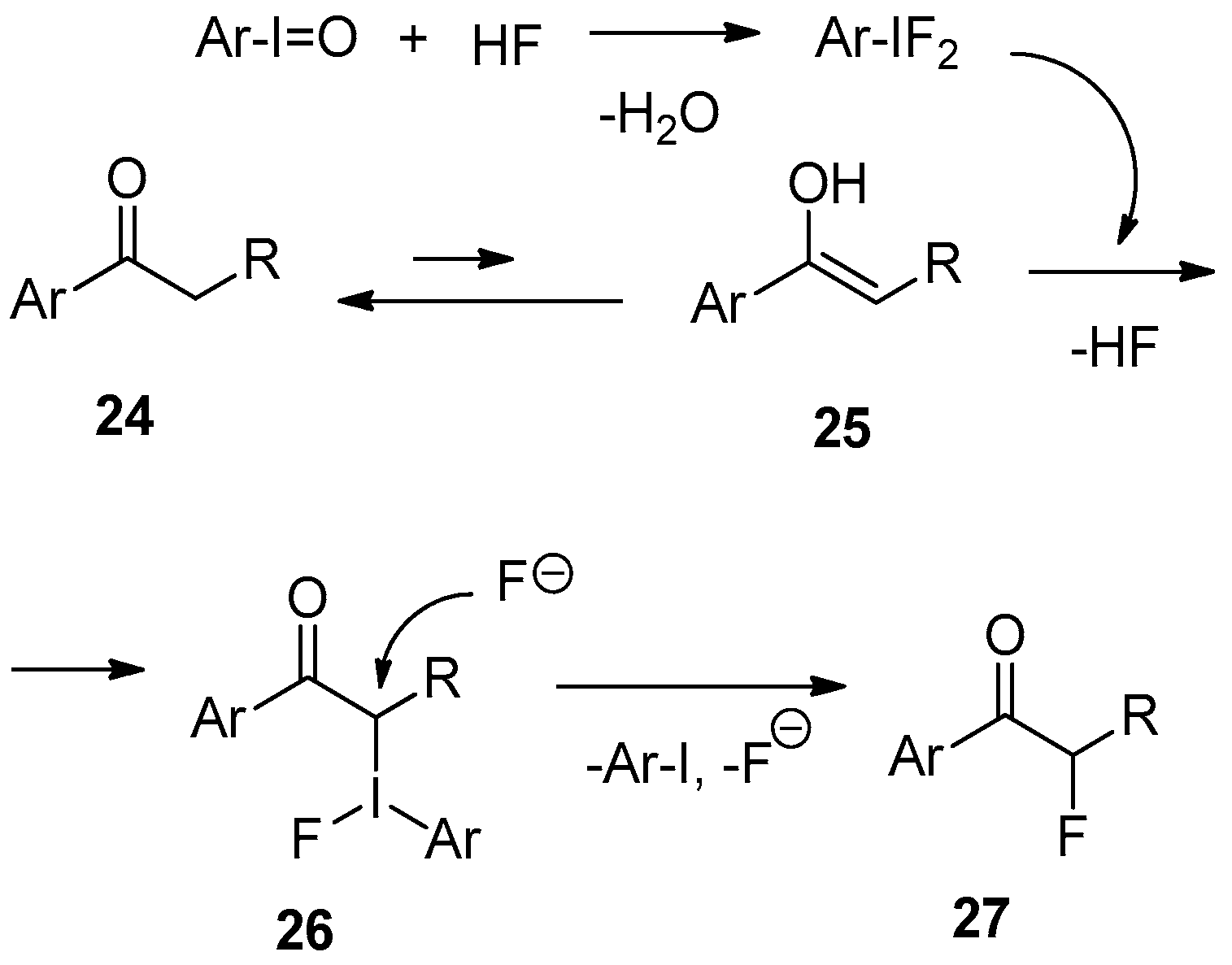
Mechanistically, the process can be envisioned as presented in Scheme 9. There are two first independent steps including in situ formation of Ar-IF2 from HF and Ar-I=O, and enolization of the starting ketone 24. Electrophilic attach of Ar-IF2 on enolate 25 results in intermediate 26, which is subjected to nucleophilic substitution with a fluoride ion giving rise to the final product 27. The net result of this reaction sequence is the “electrophilic fluorination” of 24 to 27.
4. Fluorination of Styrene Derivatives
In sharp contrast to a trifluoromethyl group, the CHF2 group is still rather scarcely represented among marketed pharmaceuticals [14,15,16,17,18,19,20,21,22]. Known biological properties of difluoromethyl-containing compounds [141], clearly suggest that the application of this fluorinated motif might be as successful as that already established for the trifluoromethylated compounds. The major reason for the current significantly lesser application of a CHF2 group in drug design is, most definitely, a lack of synthetic methods allowing for convenient installation of this functionality. Thus, most generally used methods include reaction of organozinc reagents with potassium bromodifluoroacetate [142], fluorination of gem-bistriflates and gem-dihalides [143,144,145,146], fluorodecarboxylation of dicarboxylic acids [147,148] and chlorodifluoromethylation followed by the elimination of HCl and migration of the double bond [149].
4.1. Hypervalent Iodine-Mediated Fluorination
4.1.1. Stoichiometric Hypervalent Iodine Reagents
The synthesis of difluoromethyl-containing compounds via reaction of styrene derivatives with fluorinated hypervalent iodine reagents or iodine in the presence XeF2 [150,151,152,153,154,155,156] is also a known method. However, the necessity of preparing hypervalent iodine compounds or the use of XeF2 limits its synthetic applications. With this in mind, Kitamura’s approach was examined for the fluorination of styrene substrates [157]. As presented in Scheme 10, a series of hypervalent reagents were screened and the trifluoroacetoxy derivatives were identified as the best.
It should also be noted that a separate optimization study showed that a complex of HF with pyridine was found to serve as a superior source of nucleophilic fluoride. Thus, under these optimized conditions, the target fluorinated product 28 was prepared in greater than 60% yield [157].
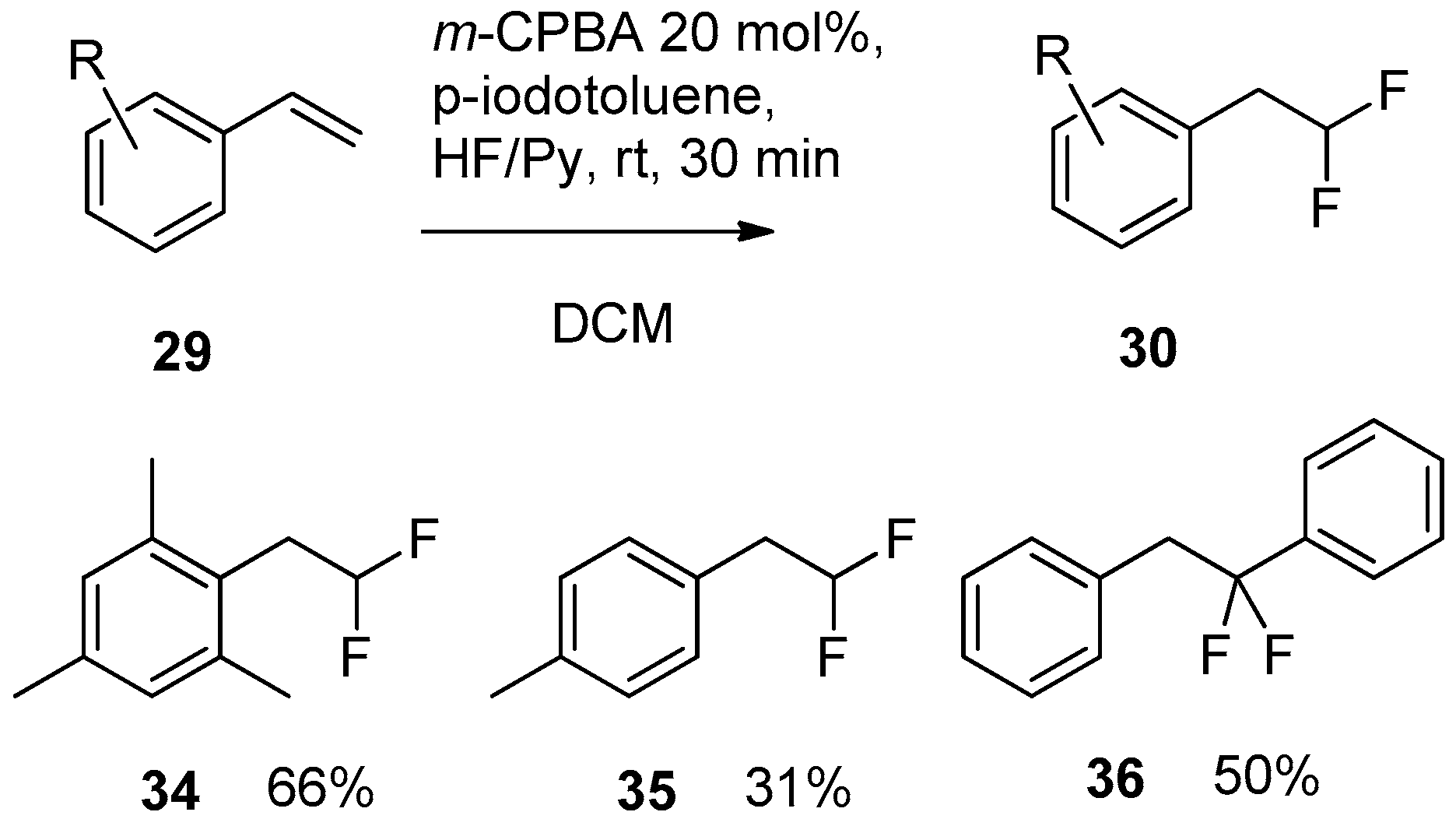
Application of these reaction conditions for fluorination of various substituted styrenes gave rather good results. This approach was applied to substrates 29 (Scheme 11) bearing alkyl, halo and OAc-type substituents in the o-, m- or p-position on the phenyl ring. The chemical yields of difluorinated products 30 ranged from 50% to 93%.
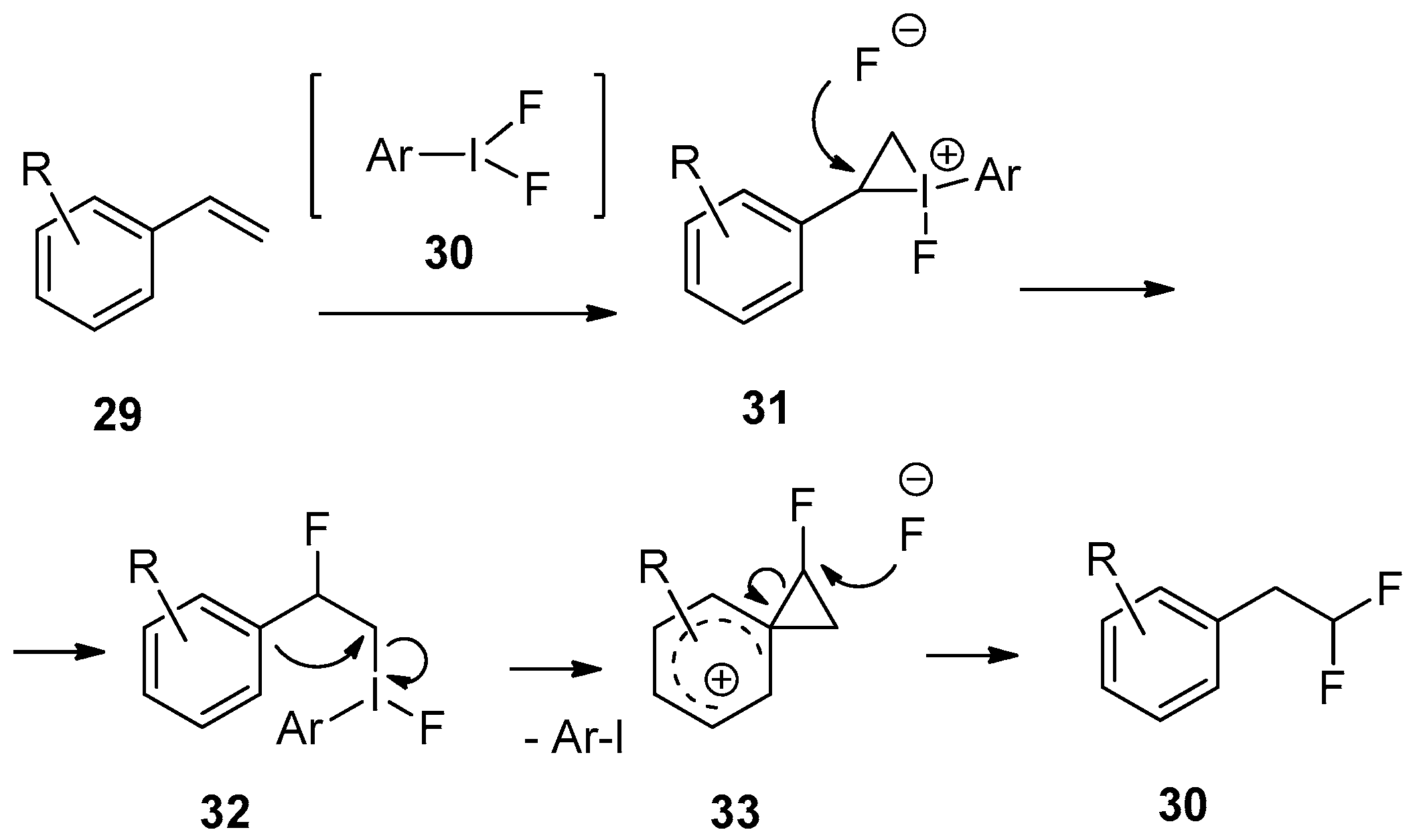
Some structural limitations of this method may arise from a mechanism involving the migration of an aryl group. As presented in Scheme 12, in situ generated electrophilic hyper-iodide reagent 30, reacts with starting styrene 29 to form three-ring intermediate 31 which is opened with fluoride to afford mono-fluorinated compound 32. The latter proceeds to a second spirocyclic three-ring intermediate 33 with the elimination of Ar-I. The final step of the process is the nucleophilic opening of 33 with fluoride to give the difluorinated product 30. One can assume that the stability of intermediate 33 will be strongly influenced by the electronic properties of the substituents on the aromatic ring. Nevertheless, as mentioned above, moderately electron-donating/withdrawing substituents, such as alkyl groups and halogens, can be tolerated giving product 30 with synthetically attractive chemical yields.
4.1.2. Catalytic Hypervalent Iodine Reactions
The catalytic version (Scheme 13) of this process was realized with application of 4-iodotoluene, HF/Py complex and m-CPBA, as the oxidizing reagent [157].
However, the outcome of the catalytic reactions was not entirely successful as difluoro products 30 were generally isolated in relatively low yields of about 50%. The highest yield (66%) in the series was obtained for the electron-rich trimethyl derivative 34. In sharp contrast, mono-methyl substituted compound 35 was prepared in only 31% yield. It is interesting to note that fluorination of 1,1-diphenylethene resulted in rearranged difluoro compound 36 isolated in 50% yield.
5. Fluorination of α,β-Unsaturated Ketones and Alcohols
One may assume that a chemically similar process can also be realized for fluorination of other types of unsaturated compounds [158]. For example, as presented in Scheme 14, it was found that α,β-unsaturated ketones of general structure 37 can be converted to ketones 38 featuring α-aryl and difluoromethyl groups [159].
Substrate generality in this reaction is rather broad as the substituent R in starting 37 can be a methyl, long-chain alkyl, tert-butyl, aromatic or heteroaromatic group. Furthermore, the aromatic ring on the carbonyl carbon can bear electron-withdrawing or -donating groups, including NO2, halogens, Alk-O and Ac-NH. The aromatic group on the unsaturated C=C fragment is migrating during the reaction to the α-position, relatively to the carbonyl, and therefore is a bit more sensitive to the nature of substitution. Nevertheless, alkyl and halogen groups on the para position of the Ar moiety seem to be perfectly tolerated [159].
A catalytic version of this process was successfully realized using m-CPBA for the in situ oxidation of p-Tol-I to p-Tol-I=O. Optimized conditions included p-Tol-I (0.2 mol%), HF/Py (40 mol%) and (1.3 mol%). Substrate generality under the catalytic conditions was not compromised, however, the chemical yields were about 5–10% lower when compared with those obtained for the reactions conducted with stoichiometric amounts of the p-Tol-I=O [159].
Of particular interest are the results reported for the fluorination of cinnamyl alcohol derivatives [160]. As presented in Scheme 15, starting compounds 39 were treated with Ph-I=O and HF/Py in dichloromethane to furnish fluorinated products 40.
It should be noted that this type of fluorination required low reaction temperature due to the sensitive nature of the cinnamyl alcohol functionality. Similar to the previously discussed fluorination of compounds with conjugated C=C bonds, the reactions occurred with migration of the Ar group. Substrate generality study of these reactions was limited to two types of products, featuring difluoromethyl 41 and difluoroethyl groups 42.
6. Cyclization–Fluorination Cascade
6.1. Homoallyl Amine Derivatives
Compounds containing the 3-fluoropyrrolidine moiety possessing a wide spectrum of biological properties. Some of them have been developed as dipeptidyl peptidase inhibitors [160,161,162,163], glucokinase activators [164] prolyl oligopeptidase inhibitors [165] and purine nucleoside phosphorylase inhibitors [166]. The most commonly used synthetic approach for preparation of 3-fluoropyrrolidines is based on fluorine substitution for hydroxy group in 3-hydroxylpyrrolidines [167,168,169,170,171]. Another approach, based on aminofluorination of alkenes [172,173,174,175,176], is more practical allowing both ring construction and introduction of a fluorine atom in one convenient synthetic sequence. It was found that the aminofluorination version of this approach can be successfully realized using the Kitamura fluorination protocol. As presented in Scheme 16, treatment of homoallyl amines 43 with a hypervalent iodine reagent and HF/Py afforded 3-fluoropyrrolidines 44 with respectable yields ranging from 50% to 87% [177].
Optimization of the reaction conditions in terms of hypervalent iodine reagent and source of HF found that a combination of HF/Py and PhI(OCOCF3)2 or PhI(OAc)2 was superior to other compounds such as PhI(OAc)2 and aqueous 55% HF and PhI(OH)OTs and HF/TEA complex [177]. As for the starting homoallyl amines, protection of the amino group with strong electron-withdrawing groups such as Ts, Ms or Ns, was found to be essential for the successful transformation. From the standpoint of generality, the process was shown to be applicable for a reasonably wide range of compounds with the substituent R bearing hydrogen or alkyl groups and with the R1 representing hydrogen, alkyl, bulky iso-alkyl and aryl groups. Quite remarkably, the reaction can be used for preparation of six-membered rings, as represented by the transformation of N-tosyl-4-pentenylamine 45 to N-tosyl-3-fluoropiperidine 46, in 69% yield [177].
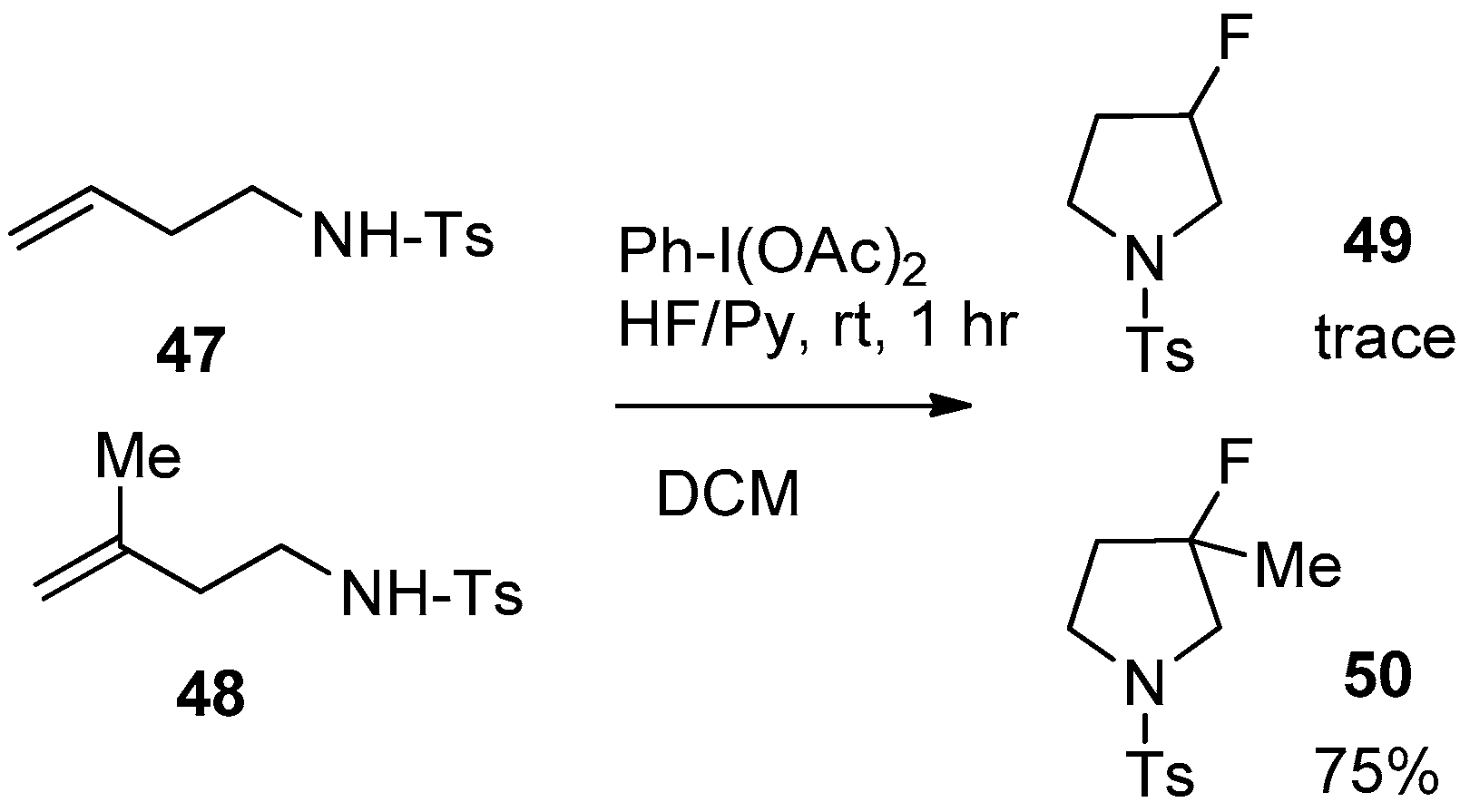
To obtain information about the reaction mechanism for this intramolecular aminofluorination, the authors conducted a competitive reaction between homoallyl amines 47 and 48 (Scheme 17).
To this end, an equimolar mixture of 47 and 48 was subjected to the aminofluorination reaction using Ph-I(OAc)2 and HF/Py. It was found that, almost exclusively, homoallyl amine 48 was transformed to 3-fluoro-3-methylpyrrolidine 50, while product 49, derived from homoallyl amine 47, was detected in the reaction mixture only in trace amounts. This result strongly suggested that the hypervalent iodine reagent preferentially reacts with the more electron-rich olefinic moiety. Based on the outcome of this competitive reaction, the authors proposed the following reaction mechanism, presented in Scheme 18.
According to the proposed mechanism, the formed in situ (difluoroiodo)benzene is activated by HF and interacts with the nucleophilic double bond of homoallyl amine 51 resulting in formation of the cyclic iodonium salt 52. The three-membered ring is opened next by the nitrogen, attacking the terminal carbon by the bridged iodonium salts 52 to afford pyrrolidine 53. The formation of intermediate 53 is followed by SN2 substitution by a fluoride ion. The final step is the deprotonation of 54 giving rise to the final 3-fluoropyrrolidine product 55.
6.2. Homoallyl Alcohol and 3-butenoic Acid Derivatives
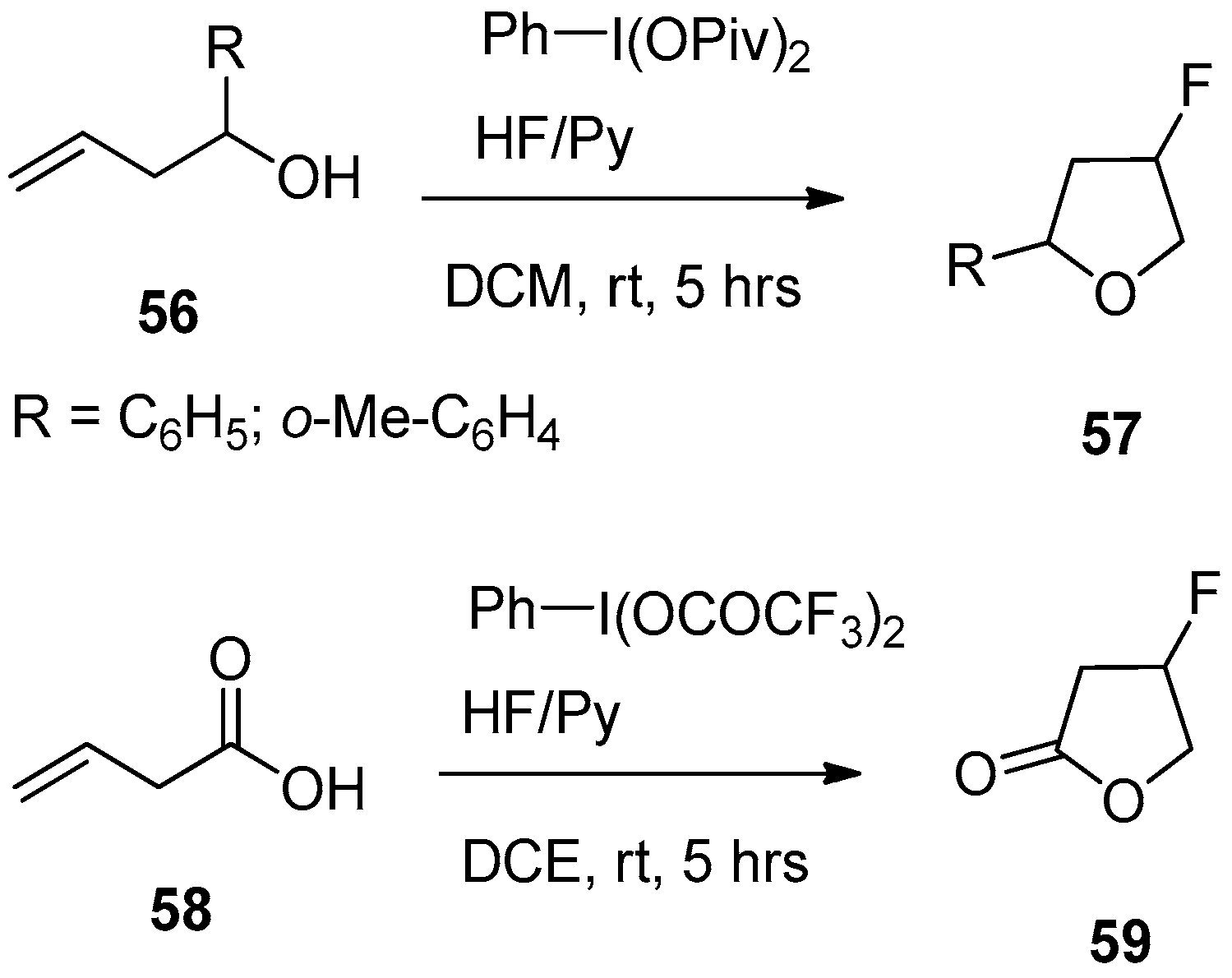
An interesting extension of this reaction cascade was demonstrated for other types of polyfunctional olefins, such as homoallyl alcohols and 3-butenoic acid derivatives [178]. The fluorination-cyclization cascade of homoallyl alcohols 56 (Scheme 19) was performed using the reagent system of PhI(OPiv)2 and HF/Py in dichloromethane. The target fluorinated tetrahydrofuran derivatives 57 were isolated with reasonably good 54−65% yields as a mixture of cis- and trans-isomers.
Under similar conditions, except using Ph-I(OCOCF3)2 as the hypervalent iodine reagent and dichloroethane as a solvent, butenoic acid 58 was transformed to fluorinated butyrolactone 59 in 45% yield. The structure of products 57 and 59 is consistent with the above-discussed mechanism (Scheme 18) for the fluorination-cyclization cascade. Thus, in the cases of homoallyl alcohols 56 and 3-butenoic acid 58 the oxygen of the hydroxy group acts as the nucleophilic element attacking the terminal carbon of the corresponding intermediate bridged iodonium salts of type 52, to complete the cyclization step.
7. Reactions with Alkynes
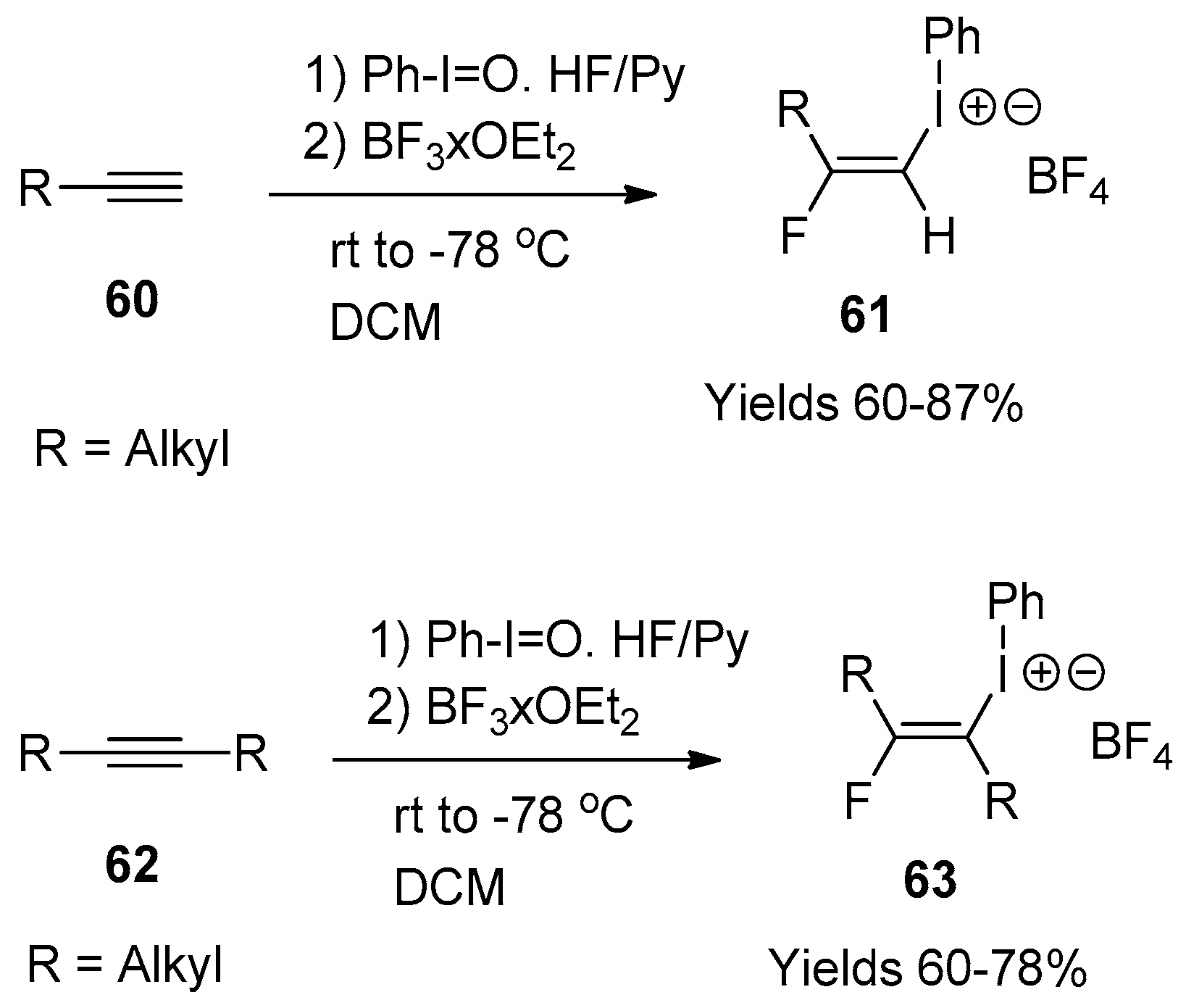
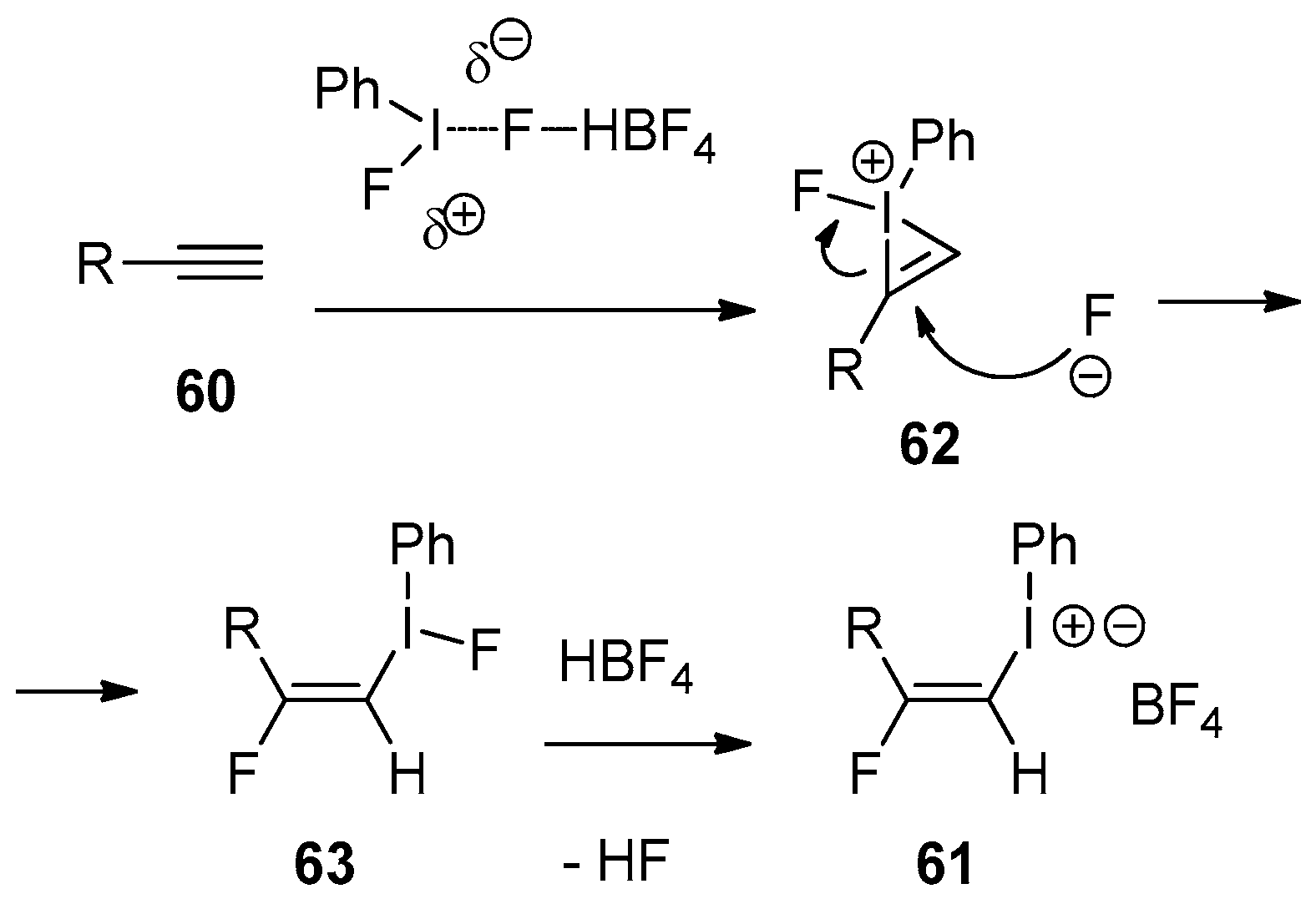
One of the most recent developments in this chemistry is the synthesis of β-fluorovinyliodonium salts via the reaction of alkynes with hypervalent iodine reagents in the presence of HF (Scheme 20) [179].
It should be noted that direct application of the conditions developed for the reactions of various C=C unsaturated compounds was found to be ineffective for alkynes. In a series of preliminary experiments, it was determined that Ph-IF2 needed some additional activation by a Lewis acid, such as BF3·OEt2. Indeed, treatment of mono-alkyl substituted alkynes 60 with Ph-I=O and HF/Py, followed by the addition of BF3·OEt2, resulted in the formation of β-fluorovinyliodonium salts 61 with a respectable chemical yield. Compound 61 is quite stable to be isolated and fully characterized. In terms of generality one can mention that the alkyl group could bear in its terminal position some functionalities, such as aromatic or heterocyclic rings, a protected alcohol or ester group. The reactions are highly regio- and stereospecific as the fluorine being added to the most substituted carbon on 60 and products 61 are obtained as trans-isomers only. This set of conditions can also be successfully applied to symmetrically disubstitute alkynes 62. The corresponding β-fluorovinyliodonium salts 63 were obtained exclusively as trans-isomers with 60–78% yields. The proposed mechanism for the reactions of Ph-I=O and HF/Py reagents with alkynes is presented in Scheme 21.
According to the proposed mechanism, the in situ generated PhIF2 is activated by HBF4 and undertakes the electrophilic addition to alkyne 60 affording bridged three-membered iodonium species 62. Intermediate 62 is subjected to the nucleophilic attack of fluoride ions leading to the formation of (E)-β-vinyliodonium fluorides 63. The final step in this sequence is the ligand exchange with HBF4 yielding final product 61. It should be emphasized that the geometric configuration of 63 is controlled by the ring-opening of iodonium species 62 with fluoride ion.
8. Conclusions
The chemistry discussed in this review article is based on the original idea of the application of HF as a source of fluorine for subsequent “electrophilic” formation of a C–F bond. The target transformation is achieved via in situ generation of the proper electrophilic species derived from hypervalent iodine compounds. The data reported so far clearly show the great synthetic value of this approach for fluorination of various 1,3-dicarbonyl compounds, aryl-alkyl ketones, styrene derivatives, α,β-unsaturated ketones and alcohols, homoallyl amine and homoallyl alcohol derivatives, 3-butenoic acids and alkynes. The major advantage of this chemistry over alternative approaches is its practicality and very attractive cost-structure, boding well for its application in large-scale synthesis of important drug intermediates or other industrial fluoro-organics. However, there are still some issues that need to be solved, to further increase the synthetic value of this methodology. One critical area of improvement would be the application of more safe and still cheaper sources of HF. Thus, most of the research has been performed using aqueous HF, and its complexes with TEA and Py. One would also suggest HF in complex with THF as an alternative source, which was reported by Professor W. R. Dolbier [179,180]. Finally, further optimization of the reaction conditions, alongside with applications of new sources of HF, leading to increased chemical yield would clearly be in the focus for future research in this exciting area of fundamental fluoro-organic methodology. As a word of caution, we would like to remind the readers that hydrogen fluoride, aqueous or in complexes with pyridine, triethylamine or in any other form, is quite toxic, highly corrosive and can easily penetrate skin and muscles destroying cell membranes and nerves. The reactions should be conducted in a well-ventilated hood using Teflon-lined reactors of tubes.
Author Contributions
Conceptualization, J.H. and V.A.S.; validation, V.A.S., G.B. and H.M.; writing—original draft preparation, G.B., H.K. and T.K.; writing—review and editing, V.A.S. and J.H.; supervision, V.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 21761132021), and IKERBASQUE, Basque Foundation for Science.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aspern, N.; Röser, S.; Rad, B.R.; Murmann, P.; Streipert, B.; Mönnighoff, X.; Tillmann, S.D.; Shevchuk, M.; Stubbmann-Kazakova, O.; Röschenthaler, G.V.; et al. Phosphorus additives for improving high voltage stability and safety of lithium ion batteries. J. Fluorine Chem. 2017, 198, 24–33. [Google Scholar] [CrossRef]
- Gschwind, F.; Rodriguez-Garcia, G.; Sandbeck, D.J.S.; Gross, A.; Weil, M.; Fichtner, M.; Hörmann, N. Fluoride ion batteries: Theoretical performance, safety, toxicity, and a combinatorial screening of new electrodes. J. Fluorine Chem. 2016, 182, 76–90. [Google Scholar] [CrossRef]
- Jeong, E.; Lee, B.H.; Doh, S.J.; Park, I.J.; Lee, Y.S. Multifunctional surface modification of an aramid fabric via direct fluorination. J. Fluorine Chem. 2012, 141, 69–75. [Google Scholar] [CrossRef]
- Simone, C.D.; Vaccaro, E.; Scola, D.A. The synthesis and characterization of highly fluorinated aromatic polyimides. J. Fluorine Chem. 2019, 224, 100–112. [Google Scholar] [CrossRef]
- Miyajima, H.; Kasuya, M.C.Z.; Hatanaka, K. New fluorous gelators for perfluorodecalin. J. Fluorine Chem. 2019. [Google Scholar] [CrossRef]
- Fersing, C.; Bouhlel, A.; Cantelli, C.; Garrigue, P.; Lisowski, V.; Guillet, B. A Comprehensive Review of Non-Covalent Radiofluorination Approaches Using Aluminum [18F] fluoride: Will [18F] AlF Replace 68Ga for Metal Chelate Labeling? Molecules 2019, 24, 2866. [Google Scholar] [CrossRef] [Green Version]
- Norton, R.S.; Leung, E.W.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of 19F-NMR in fragment-based drug discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Bailey, J.J.; Berke, S.; Schirrmacher, R. Recent advances in the development and application of radiolabeled kinase inhibitors for PET imaging. Molecules 2015, 20, 22000–22027. [Google Scholar] [CrossRef] [Green Version]
- Pretze, M.; Pietzsch, D.; Mamat, C. Recent trends in bioorthogonal click-radiolabeling reactions using fluorine-18. Molecules 2013, 18, 8618–8665. [Google Scholar] [CrossRef] [Green Version]
- De la Torre, D.G.; Albericio, F. The pharmaceutical industry in 2018. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2019, 24, 809. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluorine Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G. Chapter 4: Fluorine-Containing Agrochemicals: An Overview of Recent Developments. In Advance in Fluorine Science; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands; Volume 2, pp. 121–175. Available online: https://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S1872035806020045 (accessed on 30 April 2020).
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anticancer agents. J. Fluorine Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluorine Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluorine Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon-Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J.L. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Li, Z.; Mei, H.; Soloshonok, V.A.; Han, J.L. Detrifluoroacetylative in Situ Generated Cyclic Fluorinated Enolates for the Preparation of Compounds Featuring a C–F Stereogenic Center. ACS Omega 2019, 4, 19505–19512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, M.; Suryanarayanan, V.; Chellammal, S. A review of recent developments in the selective electrochemical fluorination of organic compounds. J. Fluorine Chem. 1997, 83, 31–40. [Google Scholar] [CrossRef]
- Wu, J. Review of recent advances in nucleophilic C–F bond-forming reactions at sp3 centers. Tetrahedron Lett. 2014, 55, 4289–4294. [Google Scholar] [CrossRef] [Green Version]
- Kuehnel, M.F.; Lentz, D.; Braun, T. Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem. Int. Ed. 2013, 52, 3328–3348. [Google Scholar] [CrossRef]
- Lectard, S.; Hamashima, Y.; Sodeoka, M. Recent Advances in Catalytic Enantioselective Fluorination Reactions. Adv. Synth. Catal. 2010, 352, 2708–2732. [Google Scholar] [CrossRef]
- Yerien, D.E.; Bonesi, S.; Postigo, A. Fluorination methods in drug discovery. Org. Biomol. Chem. 2016, 14, 8398–8427. [Google Scholar] [CrossRef] [Green Version]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic Transamination — A Metal-Free Alternative to the Reductive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth. 2011, 8, 281–294. [Google Scholar]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α,α-Disubstituted-α-Amino Acids. Synthesis 2010, 2319–2344. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sánchez-Roselló, M.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Synthesis of Fluorine Containing β-Amino Acids. Synthesis 2011, 3045–3079. [Google Scholar]
- Aceña, J.L.; Sorochinsky, A.E.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of fluorine-containing α-amino acids in enantiomerically pure form via homologation of Ni(II) complexes of glycine and alanine Schiff bases. J. Fluorine Chem. 2013, 155, 21–38. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Roeschenthaler, G.V.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Kukhar, V.P. Fluorine-containing amino acids. J. Fluorine Chem. 1994, 69, 199–205. [Google Scholar] [CrossRef]
- Furin, G.G. Some new aspects in the application of perfluoroalkyl halides in the synthesis of fluorine-containing organic compounds. Russ. Chem. Rev. 2000, 69, 491–522. [Google Scholar] [CrossRef]
- Kiss, L.; Fülöp, F. Selective Synthesis of Fluorine-Containing Cyclic β-Amino Acid Scaffolds. Chem. Rec. 2018, 18, 266–281. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Poliashko, K.O.; Kukhar, V.P.; Rozhenko, A.B.; Soloshonok, V.A.; Sorochinsky, A.E. Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun. 2012, 48, 11519–11521. [Google Scholar] [CrossRef]
- Röschenthaler, G.V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Yin, Z.; Moriwaki, H.; Abe, H.; Miwa, T.; Han, J.; Soloshonok, V.A. Large-Scale Asymmetric Synthesis of Fmoc-(S)-2-Amino-6,6,6-Trifluorohexanoic Acid. ChemistryOpen 2019, 8, 701–704. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-trifluoro-Norleucine. Symmetry 2019, 11, 578. [Google Scholar] [CrossRef] [Green Version]
- Osipov, S.N.; Kolomiets, A.F.; Bruneau, C.; Picquet, M.; Dixneuf, P.H. Synthesis of fluorine-containing cyclic amino acid derivatives via ring closing olefin metathesis. Chem. Commun. 1998, 2053–2054. [Google Scholar] [CrossRef]
- Osipov, S.N.; Artyushin, O.I.; Kolomiets, A.F.; Bruneau, C.; Picquet, M.; Dixneuf, P.H. Synthesis of Fluorine-Containing Cyclic α-Amino Acid and α-Amino Phosphonate Derivatives by Alkene Metathesis. Eur. J. Org. Chem. 2001, 3891–3897. [Google Scholar] [CrossRef]
- Tsushima, T.; Kawada, K.; Tsuji, T.; Tawara, K. Fluorine-Containing Amino Acids and Their Derivatives. 4.1 Synthesis and Antibacterial Activity of Threo and Erythro1-Fluorodehydroxylated Chloramphenicol Analogues. J. Med. Chem. 1985, 28, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Osipov, S.N.; Sewald, N.; Kolomiets, A.F.; Fokin, A.V.; Burger, K. Synthesis of α-trifluoromethyl substituted α-amino acid derivatives from methyl 3,3,3-trifluoro-2-diazopropionate. Tetrahedron Lett. 1996, 37, 615–618. [Google Scholar] [CrossRef]
- Konno, T.; Kanda, M.; Ishihara, T.; Yamanaka, H. A novel synthesis of fluorine-containing quaternary amino acid derivatives via palladium-catalyzed allylation reaction. J. Fluorine Chem. 2005, 126, 1517–1523. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Cordes, D.B.; Slawin, A.M.Z.; O’Hagan, D. Fluorine containing amino acids: Synthesis and peptide coupling of amino acids containing the all-cis tetrafluorocyclohexyl motif. Org. Biomol. Chem. 2015, 13, 5621–5624. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of Fluorine-Containing Amino Acids for Drug Design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef]
- Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. 19F NMR: A valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. [Google Scholar] [CrossRef]
- Marsh, E.N.G.; Suzuki, Y. Using 19F NMR to probe biological interactions of proteins and peptides. ACS Chem. Biol. 2014, 9, 1242–1250. [Google Scholar] [CrossRef]
- Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18F]fluoride ion. Eur. J. Org. Chem. 2008, 2008, 2853–2873. [Google Scholar] [CrossRef]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-Stage [18F]Fluorination: New Solutions to Old Problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeckel, C.; Seufert, W.; Thust, S.; Koksch, B. Evaluation of the molecular interactions of fluorinated amino acids with native polypeptides. ChemBioChem 2004, 5, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Salwiczek, M.; Koksch, B. Effects of fluorination on the folding kinetics of a heterodimeric coiled coil. ChemBioChem 2009, 10, 2867–2870. [Google Scholar] [CrossRef] [PubMed]
- Vagt, T.; Nyakatura, E.; Salwiczek, M.; Jaeckel, C.; Koksch, B. Towards identifying preferred interaction partners of fluorinated amino acids within the hydrophobic environment of a dimeric coiled coil peptide. Org. Biomol. Chem. 2010, 8, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Nyakatura, E.K.; Reimann, O.; Vagt, T.; Salwiczek, M.; Koksch, B. Accommodating fluorinated amino acids in a helical peptide environment. RSC Adv. 2013, 3, 6319–6322. [Google Scholar] [CrossRef]
- Buer, B.C.; Chugh, J.; Al-Hashimi, H.M.; Marsh, E.N.G. Using fluorine nuclear magnetic resonance to probe the interaction of membrane-active peptides with the lipid bilayer. Biochemistry 2010, 49, 5760–5765. [Google Scholar] [CrossRef]
- De Kort, D.W.; Rombouts, W.H.; Hoeben, F.J.M.; Janssen, H.M.; As, H.V.; van Duynhoven, J.P.M. Scaling behavior of dendritic nanoparticle mobility in semidilute polymer solutions. Macromolecules 2015, 48, 7585–7591. [Google Scholar] [CrossRef]
- Kubyshkin, V.; Afonin, S.; Kara, S.; Budisa, N.; Mykhailiuk, P.K.; Ulrich, A.S. (S)-Trifluoromethyl proline: Evaluation as a structural substitute of proline for solid state 19F-NMR peptide studies. Org. Biomol. Chem. 2015, 13, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.D.; Kotoris, C.C.; Hum, G. Recent advances in electrophilic fluorination. Tetrahedron 1999, 55, 12431–12477. [Google Scholar] [CrossRef]
- Szpera, R.; Moseley, D.F.J.; Smith, L.B.; Sterling, A.J.; Gouverneur, V. The Fluorination of C−H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 2019, 58, 14824–14848. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fu, L.; Chen, P.; Liu, G. Recent Advances and Perspectives of Transition Metal-Catalyzed Asymmetric Fluorination Reactions. Chin. J. Chem. 2017, 35, 1781–1788. [Google Scholar] [CrossRef]
- Kohlhepp, S.V.; Gulder, T. Hypervalent iodine(III) fluorinations of alkenes and diazo compounds: New opportunities in fluorination chemistry. Chem. Soc. Rev. 2016, 45, 6270–6288. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, T.; Revankar, H.M.; Zhang, C.P. Recent progress on fluorination in aqueous media. Green Chem. 2017, 19, 3951–3992. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Beier, P. Construction of Asymmetric Fluorinated Carbon Centers. Angew. Chem. Int. Ed. 2006, 45, 2172–2174. [Google Scholar] [CrossRef]
- Tredwell, M.; Gouverneur, V. Electrophilic fluorination of organosilanes. Org. Biomol. Chem. 2006, 4, 26–32. [Google Scholar] [CrossRef]
- Davis, F.A.; Han, W.; Murphy, C.K. Selective, electrophilic fluorinations using N-fluoro-o-benzenedisulfonimide. J. Org. Chem. 1995, 60, 4730–4737. [Google Scholar] [CrossRef]
- Differding, E.; Rüegg, G.M. Nucleophilic substitution versus electron transfer: 1. On the mechanism of electrophilic fluorinations. Tetrahedron Lett. 1991, 32, 3815–3818. [Google Scholar] [CrossRef]
- Rozatian, N.; Ashworth, I.W.; Sandford, G.; Hodgson, D.R.W. A quantitative reactivity scale for electrophilic fluorinating reagents. Chem. Sci. 2018, 9, 8692–8702. [Google Scholar] [CrossRef] [Green Version]
- Piana, S.; Devillers, I.; Togni, A.; Rothlisberger, U. The mechanism of catalytic enantioselective fluorination: Computational and experimental studies. Angew. Chem. Int. Ed. 2002, 41, 979–982. [Google Scholar] [CrossRef]
- Banks, R.E.; Mohialdin-Khaffaf, S.N.; Lal, G.S.; Sharif, I.; Syvret, R.G. 1-Alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts: A novel family of electrophilic fluorinating agents. J. Chem. Soc. Chem. Commun. 1992, 595–596. [Google Scholar] [CrossRef]
- Shibata, N. Development of shelf-stable reagents for fluoro-functionalization reactions. Bull. Chem. Soc. Jpn. 2016, 89, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N. N-Fluoro-(3,5-di-tert-butyl-4-methoxy)benzenesulfonimide (NFBSI): A sterically demanding electrophilic fluorinating reagent for enantioselective fluorination. J. Fluorine Chem. 2011, 132, 222–225. [Google Scholar] [CrossRef]
- Fukushi, K.; Suzuki, S.; Kamo, T.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Kawada, K.; Shibata, N. Methyl NFSI: Atom-economical alternative to NFSI shows higher fluorination reactivity under Lewis acid-catalysis and non-catalysis. Green Chem. 2016, 18, 1864–1868. [Google Scholar] [CrossRef]
- Fukushi, K.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Nagasaki, N.; Shibata, N. Lewis Acid-Catalyzed Selective Mono-fluorination of Malonates Using Me-NFSI. Fluorine Notes 2016, 105, 3–4. [Google Scholar]
- Zhu, C.L.; Maeno, M.; Zhang, F.G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.A.; Cahard, D. Chiral N-Fluorodibenzenesulfonimide Analogues for Enantioselective Electrophilic Fluorination and Oxidative Fluorination. Eur. J. Org. Chem. 2013, 29, 6501–6505. [Google Scholar] [CrossRef]
- Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. New approaches to enantioselective fluorination: Cinchona alkaloids combinations and chiral ligands/metal complexes. J. Fluorine Chem. 2007, 128, 469–483. [Google Scholar] [CrossRef]
- Mohar, B.; Baudoux, J.; Plaquevent, J.C.; Cahard, D. Electrophilic Fluorination Mediated by Cinchona Alkaloids: Highly Enantioselective Synthesis of α-Fluoro-α-phenylglycine Derivatives. Angew. Chem. Int. Ed. 2001, 40, 4214–4216. [Google Scholar] [CrossRef]
- Cahard, D.; Audouard, C.; Plaquevent, J.C.; Roques, N. Design, Synthesis, and Evaluation of a Novel Class of Enantioselective Electrophilic Fluorinating Agents: N-Fluoro Ammonium Salts of Cinchona Alkaloids (F-CA-BF4). Org. Lett. 2000, 2, 3699–3701. [Google Scholar] [CrossRef]
- Shibata, N.; Suzuki, E.; Takeuchi, Y. A Fundamentally New Approach to Enantioselective Fluorination Based on Cinchona Alkaloid Derivatives/Selectfluor Combination. J. Am. Chem. Soc. 2000, 122, 10728–10729. [Google Scholar] [CrossRef]
- Umemoto, T.; Nagayoshi, M. N,N′-Difluoro-1,4-diazoniabicyclo[2.2.2]octane Salts, Highly Reactive and Easy-to-Handle Electrophilic Fluorinating Agents. Bull. Chem. Soc. Jpn. 1996, 69, 2287–2295. [Google Scholar] [CrossRef]
- Kitamura, T.; Kuriki, S.; Morshed, M.H.; Hori, Y. A Practical and Convenient Fluorination of 1,3-Dicarbonyl Compounds Using Aqueous HF in the Presence of Iodosylbenzene. Org. Lett. 2011, 13, 2392–2394. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.D.; Greenhall, M.P.; Hutchinson, J. Direct fluorination of 1, 3-dicarbonyl compounds. Tetrahedron 1996, 52, 1–8. [Google Scholar] [CrossRef]
- Zajc, B.; Zupan, M. Room temperature fluorination of 1, 3-diketones and enol acetates with xenon difluoride. J. Chem. Soc. Chem. Commun. 1980, 759–760. [Google Scholar] [CrossRef]
- Yemul, S.S.; Kagan, H.B.; Setton, R. Selective fluorination by C19XeF6. Tetrahedron Lett. 1980, 21, 277–280. [Google Scholar] [CrossRef]
- Zajc, B.; Zupan, M. Fluorination with xenon difluoride. 27. The effect of catalyst on fluorination of 1,3-diketones and enol acetates. J. Org. Chem. 1982, 47, 573–575. [Google Scholar] [CrossRef]
- Inman, C.E.; Oesterling, R.E.; Tyczkowski, E.A. Reactions of Perchloryl Fluoride with Organic Compounds. II. Fluorination of Certain Active Methylene Compounds1. J. Am. Chem. Soc. 1958, 80, 6533–6535. [Google Scholar] [CrossRef]
- Machleidt, H.; Hartmann, V.I. Organische Fluorverbindungen, VI. Reaktionen cyclischer Fluor-carbonyl-Verbindungen. Liebigs Ann. Chem. 1964, 679, 9–19. [Google Scholar] [CrossRef]
- Lerman, O.; Rozen, S. Acetyl hypofluorite, a new moderating carrier of elemental fluorine and its use in fluorination of 1,3-dicarbonyl derivatives. J. Org. Chem. 1983, 48, 724–727. [Google Scholar] [CrossRef]
- Rozen, S.; Brand, M. Direct fluorination of lithium enolates with acetyl hypofluorite. Synthesis 1985, 665–667. [Google Scholar] [CrossRef]
- Rozen, S.; Hebel, D. Acyl hypofluorites. A new class of organic compounds. J. Org. Chem. 1990, 55, 2621–2623. [Google Scholar] [CrossRef]
- Rozen, S.; Menahem, Y. A novel fluorinating method for the synthesis of α-fluoroketones. Tetrahedron Lett. 1979, 20, 725–728. [Google Scholar] [CrossRef]
- Stavber, S.; Zupan, M. Room-temperature fluorination of alkenes with caesium fluoroxysulphate. J. Chem. Soc. Chem. Commun. 1981, 795–796. [Google Scholar] [CrossRef]
- Banks, R.E.; Murtagh, V.; Tsiliopoulos, E. N-halogeno compounds. Part 12 [1]. Site-specific fluorination of carbanions with perfluoro-N-fluoropiperidine [2]. J. Fluorine Chem. 1991, 52, 389–401. [Google Scholar] [CrossRef]
- Xu, Z.Q.; DesMarteau, D.D.; Gotoh, Y. N-Fluorobis [(perfluoroalkyl) sulfonyl] imides. Efficient reagents for the fluorination of 1, 3-dicarbonyl derivatives. J. Fluorine Chem. 1992, 58, 71–79. [Google Scholar] [CrossRef]
- Banks, R.E.; Lawrence, N.J.; Popplewell, A.L. Efficient electrophilic fluorination of β-dicarbonyl compounds with the selectfluor reagent F-TEDA-BF4{1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)}. J. Chem. Soc. Chem. Commun. 1994, 343–344. [Google Scholar] [CrossRef]
- Cabrera, I.; Appel, W.K. New fluorinating reagents from acesulfam sweeteners. Tetrahedron 1995, 51, 10205–10208. [Google Scholar] [CrossRef]
- Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. Synthesis, Properties, and Reactivity of N,N‘-Difluorobipyridinium and Related Salts and Their Applications as Reactive and Easy-To-Handle Electrophilic Fluorinating Agents with High Effective Fluorine Content. J. Org. Chem. 1998, 63, 3379–3385. [Google Scholar] [CrossRef]
- Frantz, R.; Hintermann, L.; Perseghini, M.; Broggini, D.; Togni, A. Titanium-catalyzed stereoselective geminal heterodihalogenation of β-ketoesters. Org. Lett. 2003, 5, 1709–1712. [Google Scholar] [CrossRef]
- Gupta, O.D.; Shreeve, J.M. Reactions of trifluoroamine oxide: A new method for selective fluorination of 1, 3-diketones and β-ketoesters. Tetrahedron Lett. 2003, 44, 2799–2801. [Google Scholar] [CrossRef]
- Stavber, G.; Zupan, M.; Stavber, S. Solvent-free fluorination of organic compounds using N–F reagents. Tetrahedron Lett. 2007, 48, 2671–2673. [Google Scholar] [CrossRef]
- Hara, S.; Sekiguchi, M.; Ohmori, A.; Fukuhara, T.; Yoneda, N. Selective fluorination of β-ketoesters using iodotoluene difluoride and a HF–amine complex. Chem. Commun. 1996, 1899–1900. [Google Scholar] [CrossRef]
- Yoshida, M.; Fujikawa, K.; Sato, S.; Hara, S. α-Fluorination of β-dicarbonyl compounds using p-iodotoluene difluoride under neutral conditions. ARKIVOC 2003, 6, 36–42. [Google Scholar]
- Gondo, K.; Kitamura, T. Reaction of iodonium ylides of 1, 3-dicarbonyl compounds with HF reagents. Molecules 2012, 17, 6625–6632. [Google Scholar] [CrossRef]
- Goudreau, S.R.; Marcoux, D.; Charette, A.B. General method for the synthesis of phenyliodonium ylides from malonate esters: Easy access to 1,1-cyclopropane diesters. J. Org. Chem. 2009, 74, 470–473. [Google Scholar] [CrossRef]
- Kitamura, T.; Kuriki, S.; Muta, K.; Morshed, M.H.; Muta, K. Facile Synthesis of 2-Fluoro-1, 3-dicarbonyl Compounds with Aqueous Hydrofluoric Acid Mediated by Iodosylarenes. Synthesis 2013, 45, 3125–3130. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Oyamada, J. A Convenient Synthesis of 2-Fluoro-and 2-Chloromalonic Esters Mediated by Hypervalent Iodine. Synthesis 2015, 47, 3241–3245. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Kuriki, S. Catalytic fluorination of 1, 3-dicarbonyl compounds using iodoarene catalysts. Tetrahedron Lett. 2013, 54, 6118–6120. [Google Scholar] [CrossRef]
- Leroy, J. 1, 2-Difluoroethylenes: Synthesis via fluoro ketones. J. Org. Chem. 1981, 46, 206–209. [Google Scholar] [CrossRef]
- Moughamir, K.; Atmani, A.; Mestdagh, H.; Rolando, C.; Francesch, C. Activation of tetrabutylammonium hydrogen difluoride with pyridine: A mild and efficient procedure for nucleophilic fluorination. Tetrahedron Lett. 1998, 39, 7305–7306. [Google Scholar] [CrossRef]
- Makosza, M.; Bujok, R. Cocatalysis in phase-transfer catalyzed fluorination of alkyl halides and sulfonates. J. Fluorine Chem. 2005, 126, 209–216. [Google Scholar] [CrossRef]
- Fuglseth, E.; Thvedt, T.H.K.; Møll, M.F.; Hoff, B.H. Electrophilic and nucleophilic side chain fluorination of para-substituted acetophenones. Tetrahedron 2008, 64, 7318–7323. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, W.; Zhen, Z.; Zou, X. One-pot α-nucleophilic fluorination of acetophenones in a deep eutectic solvent. J. Fluorine Chem. 2010, 131, 340–344. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Shang, H.; Chen, X.; Ren, X. Diastereoselective Mannich Reactions Using Fluorinated Ketones: Synthesis of Stereogenic Carbon–Fluorine Units. J. Org. Chem. 2016, 81, 9858–9866. [Google Scholar] [CrossRef]
- Xu, Y.S.; Tang, Y.; Feng, H.J.; Liu, J.T.; Hsung, R.P. A Highly Regio-and Stereoselective Synthesis of α-Fluorinated Imides via Fluorination of Chiral Enamides. Org. Lett. 2015, 17, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Saidalimu, I.; Fang, X.; Lv, W.; Yang, X.; He, X.; Zhang, J.; Wu, F. Organocatalytic Asymmetric Michael Addition/Carbon-Carbon Bond Cleavage of Trifluoromethyl α-Fluorinated gem-Diols to Nitroolefins. Adv. Synth. Catal. 2013, 355, 857–863. [Google Scholar] [CrossRef]
- Kitahara, K.; Mizutani, H.; Iwasa, S.; Shibatomi, K. Asymmetric Synthesis of α-Chloro-α-halo Ketones by Decarboxylative Chlorination of α-Halo-β-ketocarboxylic Acids. Synthesis 2019, 51, 4385–4392. [Google Scholar] [CrossRef]
- Katada, M.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Catalyst-free decarboxylative fluorination of tertiary β-keto carboxylic acids. Synlett 2018, 29, 2408–2411. [Google Scholar]
- Krespan, C.G.; Smart, B.E. Fluorocarbanion chemistry. A versatile synthesis of functionalized fluoro ketones. J. Org. Chem. 1986, 51, 320–326. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Mikami, K.; Yamazaki, T.; Welch, J.T.; Honek, J. (Eds.) Current Fluoroorganic Chemistry. New Synthetic Directions, Technologies, Materials and Biological Applications; ACS Symposium Series 949; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Xiao, X.; Xie, Y.; Su, C.; Liu, M.; Shi, Y. Organocatalytic Asymmetric Biomimetic Transamination: From α-Keto Esters to Optically Active α-Amino Acid Derivatives. J. Am. Chem. Soc. 2011, 133, 12914–12917. [Google Scholar] [CrossRef]
- Liu, M.; Li, J.; Xie, Y.; Shi, Y. An efficient synthesis of optically active trifluoromethyl aldimines via asymmetric biomimetic transamination. Chem. Commun. 2013, 49, 1404–1406. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Kukhar, V.P. Biomimetic transamination of α-keto perfluorocarboxylic esters. An efficient preparative synthesis of β,β,β-trifluoroalanine. Tetrahedron 1997, 53, 8307–8314. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally convenient asymmetric synthesis of (S)- and (R)-3-amino-4,4,4-trifluorobutanoic acid: Part I: Enantioselective biomimetic transamination of isopropyl 4,4,4-trifluoro-3-oxo-butanoate. J. Fluorine Chem. 2006, 127, 924–929. [Google Scholar] [CrossRef]
- Guo, C.; Song, J.; Gong, L.Z. Biomimetic Asymmetric 1,3-Dioplar Cycloaddition: Amino Acid Precursors in Biosynthesis Serve as Latent Azomethine Ylides. Org. Lett. 2013, 15, 2676–2679. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Soloshonok, I.V.; Kukhar, V.P.; Svedas, V.K. Biomimetic Transamination of α-Alkyl β-Keto Carboxylic Esters. Chemoenzymatic Approach to the Stereochemically Defined α-Alkyl β-Fluoroalkyl β-Amino Acids. J. Org. Chem. 1998, 63, 1878–1884. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, M.; Rong, C.; Xue, F.; Li, S.; Xie, Y.; Shi, Y. An Efficient Asymmetric Biomimetic Transamination of α-Keto Esters to Chiral α-Amino Esters. Org. Lett. 2012, 14, 5270–5273. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ono, T.; Soloshonok, I.V. Enantioselective Biomimetic Transamination of β-Keto Carboxylic Acid Derivatives. An Efficient Asymmetric Synthesis of β-Fluoroalkyl-β-Amino Acids. J. Org. Chem. 1997, 62, 7538–7539. [Google Scholar]
- Soloshonok, V.A.; Ono, T. The Effect of Substituents on the Feasibility of Azomethine-Azomethine Isomerization: New Synthetic Opportunities for Biomimetic Transamination. Tetrahedron 1996, 52, 14701–14712. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally Convenient Asymmetric Synthesis of (S)- and (R)-3-Amino-4,4,4-trifluorobutanoic Acid. Part II: Enantioselective Biomimetic Transamination of 4,4,4-Trifluoro-3-oxo-N-[(R)-1-phenylethyl)butanamide. J. Fluorine Chem. 2006, 127, 930–935. [Google Scholar] [CrossRef]
- Barnette, W.E. N-Fluoro-N-alkylsulfonamides: Useful reagents for the fluorination of carbanions. J. Am. Chem. Soc. 1984, 106, 452–454. [Google Scholar] [CrossRef]
- Differding, E.; Lang, R.W. New fluorinating reagents-I. The first enantioselective fluorination reaction. Tetrahedron Lett. 1988, 29, 6087–6090. [Google Scholar] [CrossRef]
- Resnati, G.; DesMarteau, D.D. N-fluorobis [(trifluoromethyl) sulfonyl] imide: An efficient reagent for the. alpha.-fluorination of functionalized carbonyl compounds. J. Org. Chem. 1991, 56, 4925–4929. [Google Scholar] [CrossRef]
- Davis, F.A.; Han, W. N-Fluoro-o-benzenedisulfonimide: A useful new fluorinating reagent. Tetrahedron Lett. 1991, 32, 1631–1634. [Google Scholar] [CrossRef]
- Davis, F.A.; Zhou, P.; Murphy, C.K. Asymmetric fluorination of enolates with N-fluoro 2, 10-(3, 3-dichlorocamphorsultam). Tetrahedron Lett. 1993, 34, 3971–3974. [Google Scholar] [CrossRef]
- Middleton, W.J.; Bingham, E.M. alpha-Fluorination of carbonyl compounds with trifluoromethyl hypofluorite. J. Am. Chem. Soc. 1980, 102, 4845–4846. [Google Scholar] [CrossRef]
- Purrington, S.T.; Lazaridis, N.V.; Bumgardner, C.L. Preparation of α-fluoroaldehydes and α-fluoroketones using dilute fluorine. Tetrahedron Lett. 1986, 27, 2715–2716. [Google Scholar] [CrossRef]
- Umemoto, T.; Kawada, K.; Tomita, K. N-fluoropyridinium triflate and its derivatives: Useful fluorinating agents. Tetrahedron Lett. 1986, 27. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Muta, K. Hypervalent Iodine-Promoted α-Fluorination of Acetophenone Derivatives with a Triethylamine· HF Complex. J. Org. Chem. 2014, 79, 5842–5846. [Google Scholar] [CrossRef]
- Rueeger, H.; Lueoend, R.; Rogel, O.; Rondeau, J.M.; Möbitz, H.; Machauer, R.; Jacobson, L.; Staufenbiel, M.; Desrayaud, S.; Neumann, U. Discovery of Cyclic Sulfone Hydroxyethylamines as Potent and Selective β-Site APP-Cleaving Enzyme 1 (BACE1) Inhibitors: Structure-Based Design and in Vivo Reduction of Amyloid β-Peptides. J. Med. Chem. 2012, 55, 3364–3386. [Google Scholar] [CrossRef]
- Levin, V.V.; Zemtsov, A.A.; Struchkova, M.I.; Dilman, A.D. Reactions of organozinc reagents with potassium bromodifluoroacetate. J. Fluorine Chem. 2015, 171, 97–101. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr.; Okamoto, M. Preparation of 1, 1-difluoroalkanes from aldehydes via 1, 1-bistriflates: Advantageous use of HF–Lewis base reagents. J. Fluorine Chem. 2014, 167, 96–100. [Google Scholar] [CrossRef]
- Bujok, R.; Mlkosza, M. Synthesis of 1, 1-Difluoroalkanes via Phase Transfer Catalysed Reaction of 1, 1-bis-Triflates with KF in the Presence of Cocatalyst-Ph3SnF. Synlett 2002, 1285–1286. [Google Scholar]
- Martinez, A.G.; Barcina, J.O.; Rys, A.Z.; Subramanian, L.R. A facile conversion of aliphatic aldehydes to 1, 1-difluoroalkanes. Tetrahedron Lett. 1992, 33, 7787–7788. [Google Scholar] [CrossRef]
- Bloodworth, A.J.; Bowyer, K.J.; Mitchell, J.C. A mild, convenient, halogen-exchange route to gem-difluorides and trifluorides. Tetrahedron Lett. 1987, 28, 5347–5350. [Google Scholar] [CrossRef]
- Patrick, T.B.; Johri, K.K.; White, D.H. Fluoro-decarboxylation with xenon difluoride. J. Org. Chem. 1983, 48, 4158–4159. [Google Scholar] [CrossRef]
- Patrick, T.B.; Johri, K.K.; White, D.H.; Bertrand, W.S.; Mokhtar, R.; Kilbourn, M.R.; Welch, M.J. Replacement of the carboxylic acid function with fluorine. Can. J. Chem. 1986, 64, 138–141. [Google Scholar] [CrossRef]
- Salomon, P.; Zard, S.Z. A Practical Source of Chlorodifluoromethyl Radicals. Convergent Routes to gem-Difluoroalkenes and -dienes and (2,2-Difluoroethyl)-indoles, -azaindoles, and –naphthols. Org. Lett. 2014, 16, 2926–2929. [Google Scholar] [CrossRef]
- Zupan, M.; Pollak, A. Preparation of a polymer-supported aryliodine(III) difluoride and its use to fluorinate olefins to difluorides. J. Chem. Soc. Chem. Commun. 1975, 715–716. [Google Scholar] [CrossRef]
- Carpenter, W.R. Aryliodosodifluorides. J. Org. Chem. 1966, 31, 2688–2689. [Google Scholar] [CrossRef]
- Lermontov, S.A.; Rakov, I.M.; Zefirov, N.S.; Stang, P.J. Hexafluoropropene oxide–a fluorinating reagent for the formation of element–fluorine bonds. J. Fluorine Chem. 1999, 93, 103–106. [Google Scholar] [CrossRef]
- Patrick, T.B.; Qian, S. Rearrangement of Phenylethenes on Reaction with Iodine− Xenon Difluoride. Org. Lett. 2000, 2, 3359–3360. [Google Scholar] [CrossRef]
- Hara, S.; Nakahigashi, J.; Ishi-i, K.; Fukuhara, T.; Yoneda, N. Fluorinative ring-contraction of cyclic alkenes with p-iodotoluene difluoride. Tetrahedron Lett. 1998, 39, 2589–2592. [Google Scholar] [CrossRef]
- Sawaguchi, M.; Hara, S.; Yoneda, N. Fluorination of alkenes by iodoarene difluorides. J. Fluorine Chem. 2000, 105, 313–317. [Google Scholar] [CrossRef]
- Zupan, M.; Pollak, A. Fluorination with xenon difluoride. Preparation of aryliodine/III/difluoride. J. Fluorine. Chem. 1976, 7, 445–447. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Oyamada, J. Hypervalent Iodine-Mediated Fluorination of Styrene Derivatives: Stoichiometric and Catalytic Transformation to 2,2-Difluoroethylarenes. J. Org. Chem. 2015, 80, 10431–10436. [Google Scholar] [CrossRef]
- Ilchenko, N.O.; Tasch, B.O.A.; Szabo, K.J. Mild Silver-Mediated Geminal Difluorination of Styrenes Using an Air- and Moisture-Stable Fluoroiodane Reagent. Angew. Chem. Int. Ed. 2014, 53, 12897–12901. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Yoshida, K.; Mizuno, S.; Miyake, A.; Oyamada, J. Difluorination of functionalized aromatic olefins using hypervalent iodine/HF reagents. J. Org. Chem. 2018, 83, 14853–14860. [Google Scholar] [CrossRef]
- Xu, J.; Wei, L.; Mathvink, R.; He, J.; Park, Y.J.; He, H.; Leiting, B.; Lyons, K.A.; Marsilio, F.; Patel, R.A.; et al. Discovery of potent and selective phenylalanine based dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2533–2536. [Google Scholar] [CrossRef]
- Augustyns, K.J.L.; Lambeir, A.M.; Borloo, M.; De Meester, I.; Vedernikova, I.; Vanhoof, G.; Hendriks, D.; Scharpe, S.; Haemers, A. Pyrrolidides: Synthesis and structure-activity relationship as inhibitors of dipeptidyl peptidase IV. Eur. J. Med. Chem. 1997, 32, 301–309. [Google Scholar] [CrossRef]
- Caldwell, C.G.; Chen, P.; He, J.; Parmee, E.R.; Leiting, B.; Marsilio, F.; Patel, R.A.; Wu, J.K.; Eiermann, G.J.; Petrov, A.; et al. Fluoropyrrolidine amides as dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 1265–1268. [Google Scholar] [CrossRef]
- Fukushima, H.; Hiratate, A.; Takahashi, M.; Mikami, A.; Saito-Hori, M.; Munetomo, E.; Kitano, K.; Chonan, S.; Saito, H.; Suzuki, A.; et al. Synthesis and structure–activity relationships of potent 4-fluoro-2-cyanopyrrolidine dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. 2008, 16, 4093–4106. [Google Scholar] [CrossRef]
- Mao, W.; Ning, M.; Liu, Z.; Zhu, Q.; Leng, Y.; Zhang, A. Design, synthesis, and pharmacological evaluation of benzamide derivatives as glucokinase activators. Bioorg. Med. Chem. 2012, 20, 2982–2991. [Google Scholar] [CrossRef] [PubMed]
- Goossens, F.; Vanhoof, G.; Meester, I.; Augustyns, K.; Borloo, M.; Tourwe, D.; Haemers, A.; Scharpe, S. Development and Evaluation of Peptide-Based Prolyl Oligopeptidase Inhibitors- Introduction of N-Benzyloxycarbonyl-Prolyl-3-Fluoropyrrolidine as a Lead in Inhibitor Design. Eur. J. Biochem. 1997, 250, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Murkin, A.S.; Li, L.; Schramm, V.L.; Gainsford, G.J.; Skelton, B.W. A β-Fluoroamine Inhibitor of Purine Nucleoside Phosphorylase. J. Med. Chem. 2008, 51, 5880–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardina, G.; Dondio, G.; Grugni, M. Facile and Efficient Syntheses of Novel (S)- and (R)-3-Fluoropyrrolidines and 3,3-Difluoropyrrolidine. Synlett 1995, 1995, 55–57. [Google Scholar] [CrossRef]
- Demange, L.; Cluzeau, J.; Ménez, A.; Dugave, C. Synthesis of optically pure N-Boc-protected (2R,3R)- and (2R,3S)-3-fluoroprolines. Tetrahedron Lett. 2001, 42, 651–653. [Google Scholar] [CrossRef]
- Doi, M.; Nishi, Y.; Kiritoshi, N.; Iwata, T.; Nago, M.; Nakano, H.; Uchiyama, S.; Nakazawa, T.; Wakamiya, T.; Kobayashi, Y. Simple and efficient syntheses of Boc- and Fmoc-protected 4(R)- and 4(S)-fluoroproline solely from 4(R)-hydroxyproline. Tetrahedron 2002, 58, 8453–8459. [Google Scholar] [CrossRef]
- Verniest, G.; Piron, K.; Van Hende, E.; Thuring, J.W.; Macdonald, G.; Deroose, F.; De Kimpe, N. Synthesis of aminomethylated 4-fluoropiperidines and 3-fluoropyrrolidines. Org. Biomol. Chem. 2010, 8, 2509–2512. [Google Scholar] [CrossRef]
- Fjelbye, K.; Marigo, M.; Clausen, R.P.; Juhl, K. Diastereodivergent Access to Syn and Anti 3,4-Substituted β-Fluoropyrrolidines: Enhancing or Reversing Substrate Preference. Org. Lett. 2016, 18, 1170–1173. [Google Scholar] [CrossRef]
- Kong, W.; Merino, E.; Nevado, C. Divergent Reaction Mechanisms in the Aminofluorination of Alkenes. Chimia 2014, 68, 430–435. [Google Scholar] [CrossRef]
- Chen, P.; Liu, G. Advancements in Aminofluorination of Alkenes and Alkynes: Convenient Access to β-Fluoroamines. Eur. J. Org. Chem. 2015, 2015, 4295–4309. [Google Scholar] [CrossRef]
- Combettes, L.E.; Lozano, O.; Gouverneur, V. Metal free fluoroamination of allylsilanes: A route to 3-fluoropyrrolidines. J. Fluorine Chem. 2012, 143, 167–176. [Google Scholar] [CrossRef]
- Xu, T.; Qiu, S.; Liu, G. Palladium-Catalyzed Intramolecular Aminofluorination of Styrenes. Chin. J. Chem. 2011, 29, 2785–2790. [Google Scholar] [CrossRef]
- Cui, J.; Jia, Q.; Feng, R.Z.; Liu, S.S.; He, T.; Zhang, C. Boron Trifluoride Etherate Functioning as a Fluorine Source in an Iodosobenzene-Mediated Intramolecular Aminofluorination of Homoallylic Amines. Org. Lett. 2014, 16, 1442–1445. [Google Scholar] [CrossRef]
- Kitamura, T.; Miyake, A.; Muta, K.; Oyamada, J. Hypervalent Iodine/HF Reagents for the Synthesis of 3-Fluoropyrrolidines. J. Org. Chem. 2017, 82, 11721–11726. [Google Scholar] [CrossRef]
- Kitamura, T.; Mizuno, S.; Muta, K.; Oyamada, J. Synthesis of β-Fluorovinyliodonium Salts by the Reaction of Alkynes with Hypervalent Iodine/HF Reagents. J. Org. Chem. 2018, 83, 2773–2778. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr.; Celewicz, L.; Ohnishi, K. Fluorination reactions with HF/THF medium solvolysis of N-tosyl-O-phenylhydroxylamine. Tetrahedron Lett. 1989, 30, 4929–4930. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr.; Gray, T.A.; Onnishi, K. Hydrogen Fluoride/Tetrahydrofuran as a Fluorinating Medium. A General Synthesis of 1,1,1,2-Tetrafluoro-2-alkenes. Synthesis 1987, 956–958. [Google Scholar] [CrossRef]
Figure 1.
Some commercially available N–F electrophilic fluorination reagents.

Scheme 1.
Fluorination of 3-oxo-3-phenylpropionate 5 using HF as a source of fluorine.

Scheme 2.
Mechanistic rationale of the “electrophilic” fluorination of 3-oxo-3-phenylpropionate 5 to 2-fluoro-3-oxo-3-phenylpropionate 6.
Scheme 2.
Mechanistic rationale of the “electrophilic” fluorination of 3-oxo-3-phenylpropionate 5 to 2-fluoro-3-oxo-3-phenylpropionate 6.

Scheme 3.
Synthesis of iodonium ylide 11 and its reactions with HF and HCl.

Scheme 4.
General synthetic applications of HF/PhIO for “electrophilic” fluorination of 1,3-dicarbonyl compounds.
Scheme 4.
General synthetic applications of HF/PhIO for “electrophilic” fluorination of 1,3-dicarbonyl compounds.

Scheme 5.
Proposed catalytic cycle of the iodoarene-mediated “electrophilic” fluorination.

Scheme 6.
“Electrophilic” fluorination using a catalytic amount of Ar-I.

Scheme 7.
Fluorination of acetophenone.

Scheme 8.
Substrate generality of the fluorination of ketones.

Scheme 9.
Proposed mechanism for electrophilic fluorination.

Scheme 10.
Fluorination of styrene.

Scheme 11.
Generality of styrene fluorination using hypervalent iodine and HF complex with pyridine.
Scheme 11.
Generality of styrene fluorination using hypervalent iodine and HF complex with pyridine.

Scheme 12.
Mechanism of styrene fluorination.

Scheme 13.
Catalytic version of styrene fluorination.

Scheme 14.
Fluorination of α,β-unsaturated ketones.

Scheme 15.
Fluorination of cinnamyl alcohol derivatives.

Scheme 16.
Intramolecular aminofluorination of homoallyl amine via cyclization-fluorination cascade.
Scheme 16.
Intramolecular aminofluorination of homoallyl amine via cyclization-fluorination cascade.

Scheme 17.
Competitive intramolecular aminofluorination.

Scheme 18.
Mechanism of the intramolecular aminofluorination.

Scheme 19.
Fluorination of homoallyl alcohols and 3-butenoic acid derivatives.

Scheme 20.
Synthesis of β-fluorovinyliodonium salts by the reaction of alkynes with hypervalent iodine/HF reagent.
Scheme 20.
Synthesis of β-fluorovinyliodonium salts by the reaction of alkynes with hypervalent iodine/HF reagent.

Scheme 21.
Proposed mechanism for the reactions of alkynes with Ph-I=O and HF/Py in the presence of BF3·OEt2.
Scheme 21.
Proposed mechanism for the reactions of alkynes with Ph-I=O and HF/Py in the presence of BF3·OEt2.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, J.; Butler, G.; Moriwaki, H.; Konno, H.; Soloshonok, V.A.; Kitamura, T. Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine. Molecules 2020, 25, 2116. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092116
AMA Style
Han J, Butler G, Moriwaki H, Konno H, Soloshonok VA, Kitamura T. Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine. Molecules. 2020; 25(9):2116. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092116
Chicago/Turabian StyleHan, Jianlin, Greg Butler, Hiroki Moriwaki, Hiroyuki Konno, Vadim A. Soloshonok, and Tsugio Kitamura. 2020. "Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine" Molecules 25, no. 9: 2116. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092116