
Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
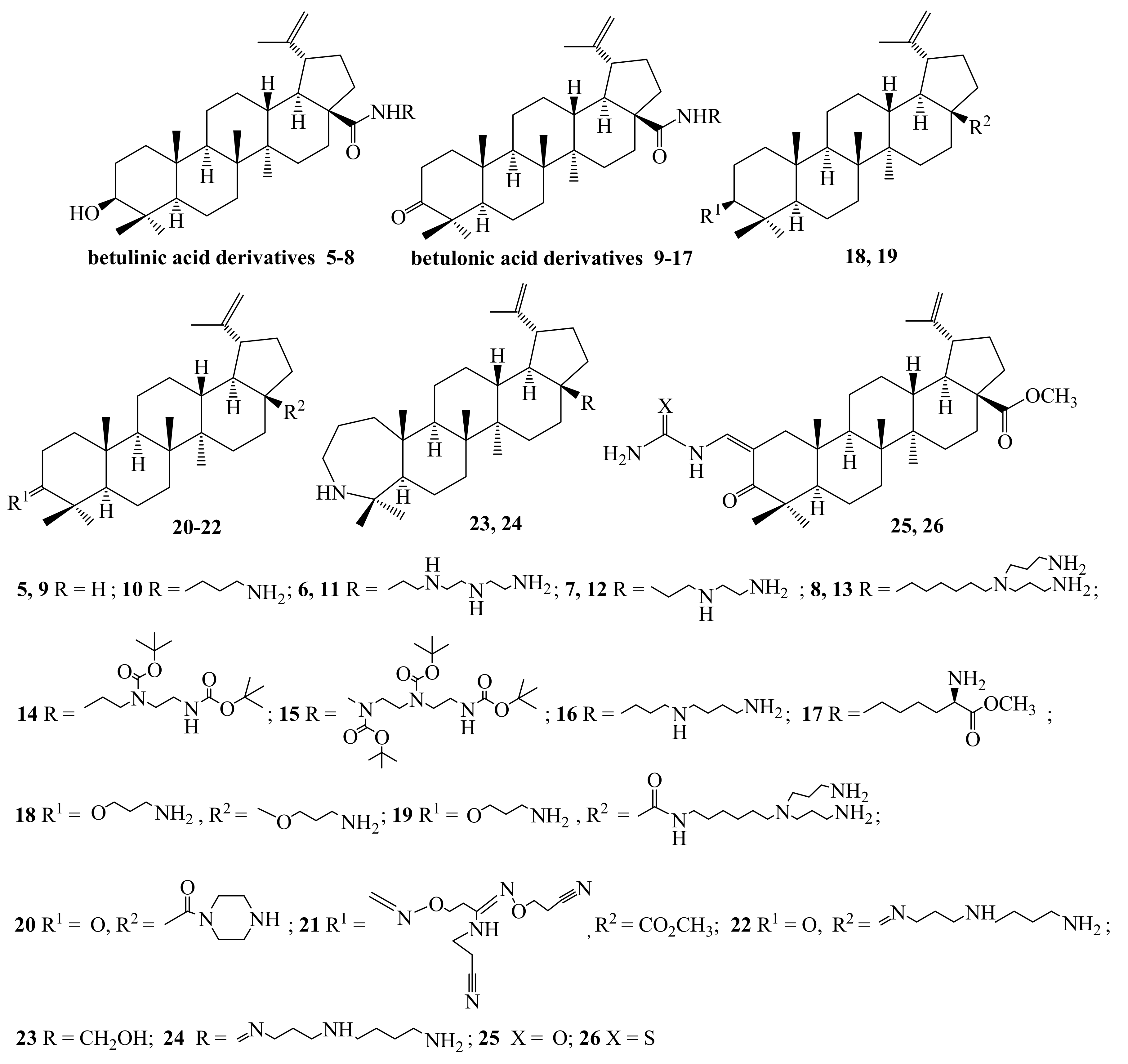
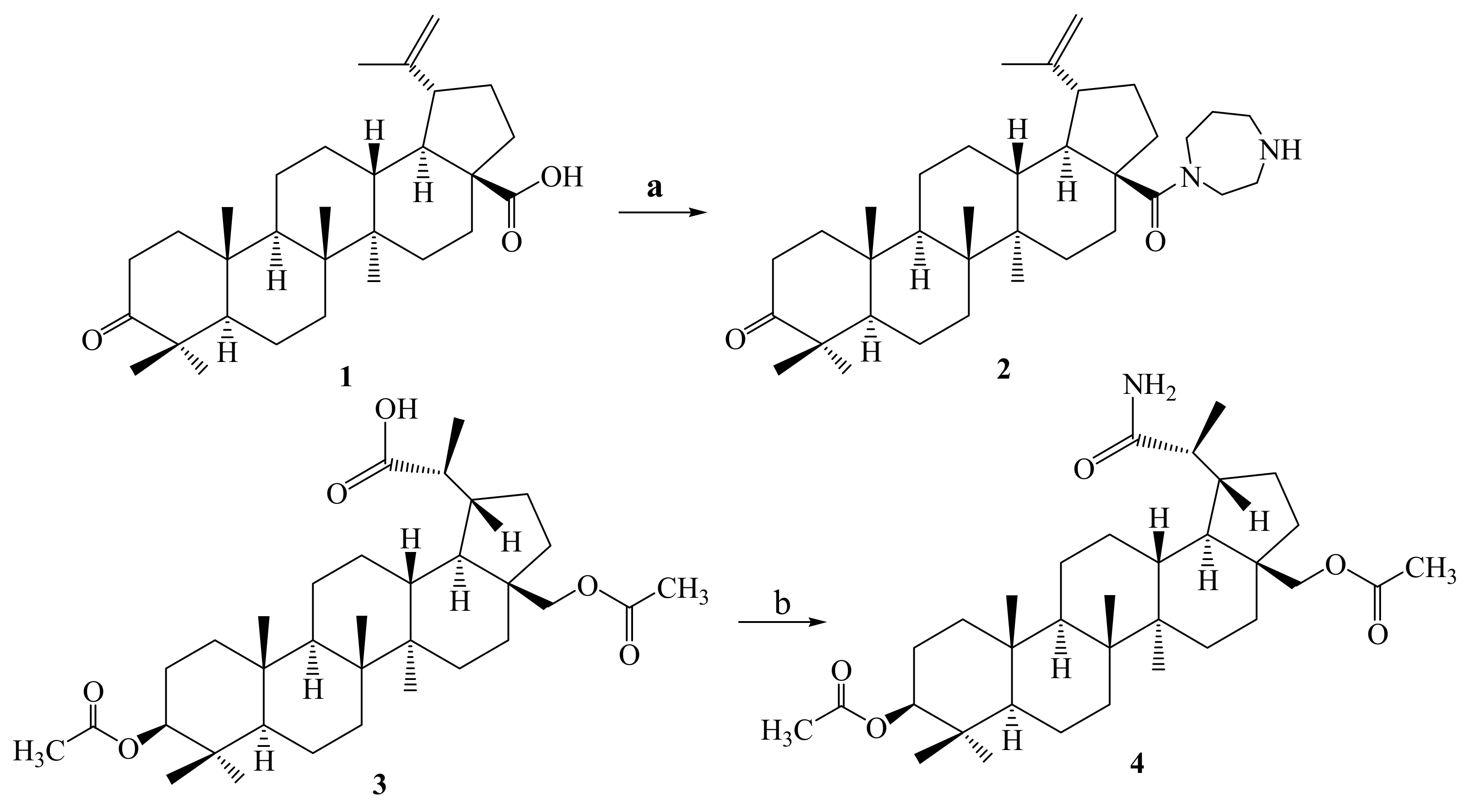
2.1. Chemistry
2.2. Biological Evaluation
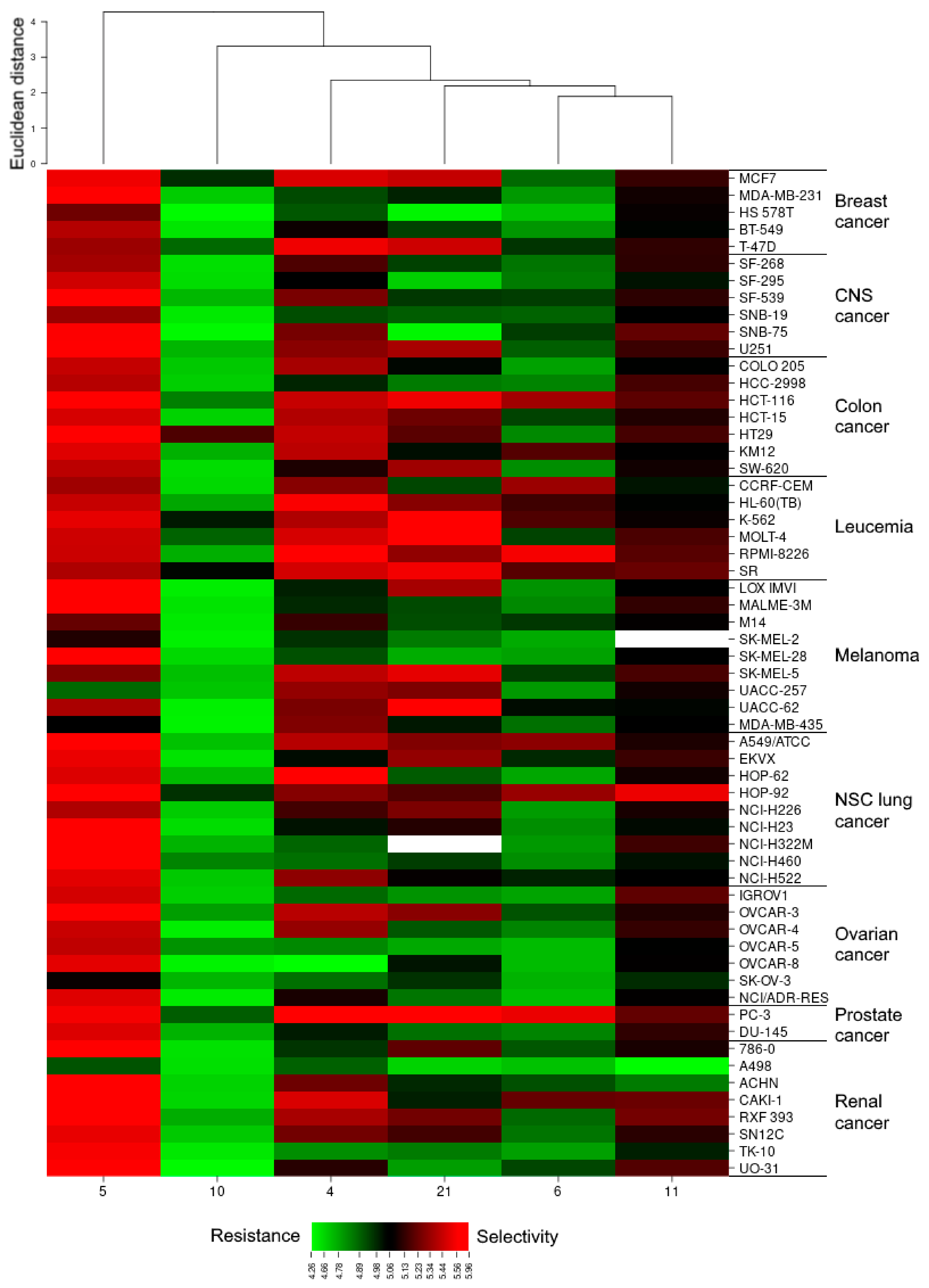
2.2.1. Evaluation of In Vitro Antiproliferative Activity by NCI
2.2.2. CellMinerTM and Gene Enrichment Analysis
2.2.3. α-Glucosidase Inhibition
2.2.4. Antibacterial and Fungicidal Activities
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis of 28-(1,4-Diazepan-1-yl)-28-oxolup-20(29)-en-3-one 2
3.1.3. Synthesis of 3β,28-Diacetyloxy-(20R)-29-amino-29-oxolupane 4
3.2. Pharmacological Studies
3.2.1. In Vitro Cancer Screen in NCI, USA
3.2.2. CellMiner and Gene Ontology Enrichment Analysis
3.2.3. Yeast α-Glucosidase Activity Assay
3.2.4. Endoplasmic Reticulum α-Glucosidase Activity Assay
3.2.5. Antibacterial and Antifungal Assays
Antibacterial Assay
Antifungal Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kvasnica, M.; Urban, M.N.; Dickinson, J.; Sarek, J. Pentacyclic triterpenoids with nitrogen- and sulfur-containing heterocycles: Synthesis and medicinal significance. Nat. Prod. Rep. 2015, 32, 1303–1330. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Leal, A.S.; Valdeira, A.S.; Gonçalves, B.M.F.; Alho, D.P.S.; Figueiredo, S.A.C.; Silvestre, S.M.; Mendes, V.I.S. Oleanane-, ursane-, and quinone methide friedelane-type triterpenoid derivatives: Recent advances in cancer treatment. Eur. J. Med. Chem. 2017, 142, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.L.C.; Freire, C.S.R.; Silvestre, A.J.D.; Silva, A.M.S. Recent Developments in the Functionalization of Betulinic Acid and Its Natural Analogues: A Route to New Bioactive Compounds. Molecules 2019, 24, 355. [Google Scholar] [CrossRef] [PubMed]
- Bildziukevich, U.; Kaletova, E.; Saman, D.; Sievanen, E.; Kolehmainen, E.T.; Slouf, M.; Wimmer, Z. Spectral and microscopic study of self-assambly of novel cationic spermine amides of betulinic acid. Steroids 2017, 117, 90–96. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Rarova, L.; Janovska, L.; Saman, D.; Wimmer, Z. Enhancing effect of cystamine in its amides with betulinic acid as antimicrobial and antitumor agent in vitro. Steroids 2019, 148, 91–98. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Brunel, J.M.; Khusnutdinova, E.F.; Negrel, S.; Giniyatullina, G.V.; Lopatina, T.V.; Petrova, A.V. A-ring modified triterpenoids and their spermidine-aldimines with strong antibacterial activity. Molbank 2019, M1078. [Google Scholar] [CrossRef] [Green Version]
- Khusnutdinova, E.F.; Apryshko, G.N.; Petrova, A.V.; Kukovinets, O.S.; Kazakova, O.B. The synthesis and selective cytotoxicity of new Mannich bases derivatives of 19- and 28-alkynyltriterpenoids. Russ. J. Org. Chem. 2018, 1, 123–127. [Google Scholar] [CrossRef]
- Kahnt, M.; Hoenke, S.; Fischer, L.; Al-Harrasi, A.; Csuk, R. Synthesis and cytotoxicity evaluation of DOTA-conjugates of ursolic acid. Molecules 2019, 24, E2254. [Google Scholar] [CrossRef] [Green Version]
- Medvedeva, N.I.; Kazakova, O.B.; Lopatina, T.V.; Smirnova, I.E.; Giniyatullina, G.V.; Baikova, I.P.; Kataev, V.E. Synthesis and antimycobacterial activity of triterpenic A-ring azepanes. Eur. J. Med. Chem. 2018, 143, 464–472. [Google Scholar] [CrossRef]
- Smirnova, I.E.; Kazakova, O.B. Structure—Anti-influenza Type a Activity Relationship among a Series of Nitrogen Lupane Triterpenoids. Nat. Prod. Commun. 2018, 13, 1267–1270. [Google Scholar] [CrossRef] [Green Version]
- Spivak, A.Y.; Khalitova, R.R.; Nedopekina, D.A.; Gubaidullin, R.R. Antimicrobial properties of amine- and guanidine-functionalized derivatives of betulinic, ursolic and oleanolic acids: Synthesis and structure/activity evaluation. Steroids 2020, 154, 108530. [Google Scholar] [CrossRef]
- Savage, P. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol. Lett. 2002, 217, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sills, A.K., Jr.; Williams, J.I.; Tyler, B.M.; Epstein, D.S.; Sipos, E.P.; Davis, J.D.; McLane, M.P.; Pitchford, S.; Cheshire, K.; Gannon, F.H.; et al. Squalamine inhibits angiogenesis and solid tumor growth in vivo and perturbs embryonic vasculature. Cancer Res. 1998, 58, 2784–2792. [Google Scholar] [PubMed]
- Moore, K.S.; Wehrli, S.; Roder, H.; Rogers, M.; Forrest, J.N.; McCrimmon, D.; Zasloff, M. Squalamine: An aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. USA 1993, 90, 1354–1358. [Google Scholar] [CrossRef] [Green Version]
- Lantz, K.A.; Hart, S.G.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by Trodusquemine (MSI-1436) Causes Fat-specific Weight Loss in Diet-induced Obese Mice. Obes. Silver Spring 2010, 18, 1516–1523. [Google Scholar] [CrossRef]
- Borselli, D.; Lieutaud, A.; Thefenne, H.; Garnotel, E.; Pagès, J.M.; Brunel, J.M.; Bolla, J.M. Polyamino-isoprenic derivatives block intrinsic resistance of P. aeruginosa to doxycycline and chloramphenicol in vitro. PLoS ONE 2016, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Giniyatullina, G.V.; Flekhter, O.B.; Tolstikov, G.A. Synthesis of squalamine analogues on the basis of lupane triterpenoids. Mendeleev Commun. 2009, 19, 32–33. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Kazakova, O.B.; Medvedeva, N.I.; Sorokina, I.V.; Zhukova, N.A.; Tolstikova, T.G.; Tolstikov, G.A. Synthesis of aminopropylamino derivatives of betulinic and oleanolic acids. Russ. J. Bioog. Chem. 2013, 39, 329–337. [Google Scholar] [CrossRef]
- Giniyatyllina, G.V.; Smirnova, I.E.; Kazakova, O.B.; Yavorskaya, N.P.; Golubeva, I.S.; Zhukova, O.S.; Pugacheva, R.B.; Apryshko, G.N.; Poroikov, V.V. Synthesis and anticancer activity of aminopropoxytriterpenoids. Med. Chem. Res. 2015, 24, 3423–3436. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Tolstikov, G.A. Synthesis of a-secomethylenamino- and substituted amidoximotriterpenoids. Russ. J. Bioog. Chem. 2011, 37, 619–625. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Medvedeva, N.I.; Tolstikov, G.A. Synthesis of a triterpene-spermidine conjugate. Russ. J. Org. Chem. 2012, 48, 1366–1369. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Malík, M.; Özdemir, Z.; Rárová, L.; Janovská, L.; Šlouf, M.; Šaman, D.; Šarek, J.; Nonappa; Wimmer, Z. Spermine amides of selected triterpenoid acids: Dynamic supramolecular system formation influences the cytotoxicity of the drugs. J. Mater. Chem. B 2020, 8, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Giniyatullina, G.V.; Kazakova, O.B.; Salimova, E.V.; Tolstikov, G.A. Synthesis of new betulonic and oleanonic acid amides. Chem. Nat. Comp. 2011, 47, 68–72. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Tolstikov, G.A.; Baikova, I.P.; Zaprutko, L.; Apryshko, G.N. Synthesis and antitumor activity of aminopropoxy derivatives of betulin, erythrodiol, and uvaol. Rus. J. Bioorg. Chem. 2011, 37, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.B.; Medvedeva, N.I.; Suponitskii, K.Y. Synthesis and molecular structure of 3β,28-diacetoxy-(20R)-lupan-29-oic acid. Chem. Nat. Comp. 2012, 41, 83–85. [Google Scholar] [CrossRef]
- Wiemann, J.; Heller, L.; Perl, V.; Kluge, R.; Ströhl, D.; Csuk, R. Betulinic acid derived hydroxamates and betulin derived carbamates are interesting scaffolds for the synthesis of novel cytotoxic compounds. Eur. J. Med. Chem. 2015, 106, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Ganguly, A.; Chowdhuri, S.; Yousuf, M.; Ghosh, A.; Barui, A.K.; Kotcherlakota, R.; Adhikari, S.; Banerjee, R. Bis-Arylidene Oxindole–Betulinic Acid Conjugate: A Fluorescent Cancer Cell Detector with Potent Anticancer Activity. ACS Med. Chem. Lett. 2015, 6, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Alakurtti, S.; Yli-Kauhaluoma, J.; Mäkelä, T.; Koskimies, S.; Bergström, S.; Hokkanen, H.; Menzler-Hokkanen, I. Betulin Derived Compounds as Anti-Feedants for Plant Pests. U.S. Patent 20120035224A1, 9 February 2012. [Google Scholar]
- Grever, M.R.; Schepartz, S.A.; Chabner, B.A. The National Cancer Institute: Cancer drug discovery and development program. Semin. Oncol. 1992, 19, 622–638. [Google Scholar]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Rev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Jangley, J.; Cronisie, P.; Viagro-Wolff, A.; et al. Feasibility of a highflux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Acton, E.M.; Narayanan, V.L.; Risbood, P.A.; Shoemaker, R.H.; Vistica, D.T.; Boyd, M.R. Anticancer Specificity of Some Ellipticinium Salts against Human Brain Tumors in vitro. J. Med. Chem. 1994, 37, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J.; Insuasty, B. Synthesis and in Vitro Antitumor Activity of a Novel Series of 2-Pyrazoline Derivatives Bearing the 4-Aryloxy-7-chloroquinoline Fragment. Molecules 2014, 19, 18656–18675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer Ther. 2007, 6, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Konopleva, M.; Hail, N.; Shi, Y.X.; McQueen, T.; Hsu, T.; Evans, R.; Honda, T.; Gribble, G.W.; Sporn, M.; et al. 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J. Biol. Chem. 2005, 280, 36273–36282. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Siegel, D.; Vogl, D.T.; Stinnett, J.; Narayanan, G.; Bapsy, P.P.; Ganesan, T.; Jain, M.; Prasad, S.V.S.; McNair, D.S.; et al. A Phase 1 Trial of Fluphenazine HCl (Fz), a Serotonin Antagonist, in Relapsed and Refractory Multiple Myeloma. Blood 2008, 112, 5188. [Google Scholar] [CrossRef]
- Di Carlo, F.; Conti, G.; Reboani, C. Interference of gestagens and androgens with rat uterine oestrogen receptors. J. Endocrinol. 1978, 77, 49–55. [Google Scholar] [CrossRef]
- Rocha, W.; Sanchez, R.; Deschênes, J.; Auger, A.; Hébert, E.; White, J.H.; Mader, S. Opposite Effects of Histone Deacetylase Inhibitors on Glucocorticoid and Estrogen Signaling in Human Endometrial Ishikawa Cells. Mol. Pharmacol. 2005, 68, 1852–1862. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, G.; Giunchi, L.; Ronchetti, S.; Krausz, L.T.; Bartoli, A.; Moraca, R.; Migliorati, G.; Riccardi, C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6216–6221. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, Z.; Mahesh, S.P.; Pantanelli, S.; Hwang, F.S.; Siu, W.O.; Nussenblatt, R.B. Glucocorticoid-induced Tumor Necrosis Factor Receptor Negatively Regulates Activation of Human Primary Natural Killer (NK) Cells by Blocking Proliferative Signals and Increasing NK Cell Apoptosis. J. Biol. Chem. 2008, 283, 8202–8210. [Google Scholar] [CrossRef] [Green Version]
- Moroz, A.; Eppolito, C.; Li, Q.; Tao, J.; Clegg, C.H.; Shrikant, P.A. IL-21 Enhances and Sustains CD8 + T Cell Responses to Achieve Durable Tumor Immunity: Comparative Evaluation of IL-2, IL-15, and IL-21. J. Immunol. 2004, 173, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Inoue, J.; Imoto, I.; Otsuki, T.; Iida, S.; Ueda, R.; Inazawa, J. POU2AF1, an amplification target at 11q23, promotes growth of multiple myeloma cells by directly regulating expression of a B-cell maturation factor, TNFRSF17. Oncogene 2008, 27, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Chapellier, M.; Peña-Martínez, P.; Ramakrishnan, R.; Eriksson, M.; Talkhoncheh, M.S.; Orsmark-Pietras, C.; Lilljebjörn, H.; Högberg, C.; Hagström-Andersson, A.; Fioretos, T.; et al. Arrayed molecular barcoding identifies TNFSF13 as a positive regulator of acute myeloid leukemia-initiating cells. Haematologica 2019, 104, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Lundin, M.; Baltscheffsky, H.; Ronne, H. Yeast PPA2 gene encodes a mitochondrial inorganic pyrophosphatase that is essential for mitochondrial function. J. Biol. Chem. 1991, 266, 12168–12172. [Google Scholar] [PubMed]
- Zewail, A.; Xie, M.W.; Xing, Y.; Lin, L.; Zhang, P.F.; Zou, W.; Saxe, J.P.; Huang, J. Novel functions of the phosphatidylinositol metabolic pathway discovered by a chemical genomics screen with wortmannin. Proc. Natl. Acad. Sci. USA 2003, 100, 3345–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.Z.; Akter, R.; Kleve, M.G.; Gealt, M.A. Wortmannin induces MCF-7 breast cancer cell death via the apoptotic pathway, involving chromatin condensation, generation of reactive oxygen species, and membrane blebbing. Breast Cancer Targets Ther. 2012, 4, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Blommaart, E.F.C.; Krause, U.; Schellens, J.P.M.; Vreeling-Sindelarova, H.; Meijer, A.J. The Phosphatidylinositol 3-Kinase Inhibitors Wortmannin and LY294002 Inhibit Autophagy in Isolated Rat Hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Pietras, R.J.; Marquez-Garban, D.C. Membrane-Associated Estrogen Receptor Signaling Pathways in Human Cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [Green Version]
- Bennett, L.L.; Smithers, D.; Rose, L.M.; Adamson, D.J.; Thomas, H.J. Inhibition of synthesis of pyrimidine nucleotides by 2-hydroxy-3-(3,3-dichloroallyl)-1,4-naphthoquinone. Cancer Res. 1979, 39, 4868–4874. [Google Scholar]
- Shah, N.; Thomas, T.J.; Lewis, J.S.; Klinge, C.M.; Shirahata, A.; Gelinas, C.; Thomas, T. Regulation of estrogenic and nuclear factor κB functions by polyamines and their role in polyamine analog-induced apoptosis of breast cancer cells. Oncogene 2001, 20, 1715–1729. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Kulkarni, G.D.; Gallo, M.A.; Greenfield, N.; Lewis, J.S.; Shirahata, A.; Thomas, T.J. Effects of natural and synthetic polyamines on the conformation of an oligodeoxyribonucleotide with the estrogen response element. Nucl. Acids Res. 1997, 25, 2396–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayanathan, V.; Thomas, T.J.; Nair, S.K.; Shirahata, A.; Gallo, M.A.; Thomas, T. Bending of the estrogen response element by polyamines and estrogen receptors α and β: A fluorescence resonance energy transfer study. Int. J. Biochem. Cell Biol. 2006, 38, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xing, Y.; Wen, C.; Yu, X.; Sun, W.; Xiu, Z.; Dong, Y. Pentacyclic triterpenes as α-glucosidase and α-amylase inhibitors: Structure-activity relationships and the synergism with acarbose. Bioorg. Med. Chem. Lett. 2017, 27, 5065–5070. [Google Scholar] [CrossRef]
- Olennikov, D.N.; Gornostai, T.G.; Penzina, T.A.; Borovskii, G.B. Lupane triterpenoids and sterols from Inonotus rheades Mycelium and their anti-glucosidase activity. Chem. Nat. Compd. 2017, 53, 988–990. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Thu, H.N.T.; Tu, A.L.T.; Thanh, T.N.; Thi, C.B.; Babkov, D.A.; Kazakova, O.B. Structural modifications of 2,3-indolobetulinic acid: Design and synthesis of highly potent α-glucosidase inhibitors. Bioorg. Chem. 2019, 88, 102957. [Google Scholar] [CrossRef] [PubMed]
- Gundoju, N.; Bokam, R.; Yalavarthi, N.R.; Azad, R.; Ponnapalli, M.G. Betulinic acid derivatives: A new class of α-glucosidase inhibitors and LPS-stimulated nitric oxide production inhibition on mouse macrophage RAW 264.7 cells. Nat. Prod. Res. 2019, 33, 2618–2622. [Google Scholar] [CrossRef]
- Ouyang, J.-K.; Dong, L.-M.; Xu, Q.-L.; Wang, J.; Liu, S.-B.; Qian, T.; Yuan, Y.-F.; Tan, J.-W. Triterpenoids with α-glucosidase inhibitory activity and cytotoxic activity from the leaves of Akebia trifoliate. RSC Adv. 2018, 8, 40483–40489. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.P.; Le, T.D.; Phan, N.M.; Bui, T.D.; Mai, D.T. Triterpene saponins with α-glucosidase inhibition and cytotoxic activity from the leaves of Schefflera sessiliflora. J. Asian Nat. Prod. Res. 2016, 18, 542–550. [Google Scholar] [CrossRef]
- Gerber-Lemaire, S.; Juillerat-Jeanneret, L. Glycosylation Pathways as Drug Targets for Cancer: Glycosidase Inhibitors. Mini-Rev. Med. Chem. 2006, 6, 104–1052. [Google Scholar] [CrossRef]
- Liu, X.; Zang, X.; Yin, X.; Yang, W.; Huang, J.; Huang, J.; Yu, C.; Ke, C.; Hong, Y. Semi-synthesis of C28-modified triterpene acid derivatives from maslinic acid or corosolic acid as potential α-glucosidase inhibitors. Bioorg. Chem. 2020, 97, 103694. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Chang, J.; Partis, R.A.; Mueller, R.A.; Chrest, F.J.; Passaniti, A. The α-Glucosidase I Inhibitor Castanospermine Alters Endothelial Cell Glycosylation, Prevents Angiogenesis, and Inhibits Tumor Growth. Cancer Res. 1995, 55, 2920. [Google Scholar] [PubMed]
- Teicher, B.A.; Williams, J.I.; Takeuchi, H.; Ara, G.; Herbst, R.S.; Buxton, D. Potential of the aminosterol, squalamine in combination therapy in the rat 13,762 mammary carcinoma and the murine Lewis lung carcinoma. Anticancer Res. 1998, 18, 2567. [Google Scholar]
- Akhter, S.; Nath, S.K.; Tse, G.M.; Williams, J.; Zasloff, M.; Donowitz, M. Squalamine, a novel cationic steroid, specifically inhibits the brush- border Na+/H+ exchanger isoform NHE3. Am. J. Physiol. Cell Physiol. 1999, 276, C136–C144. [Google Scholar] [CrossRef]
- Hiramatsu, R.; Fukuhara, S.; Mitsuda, S.; Yokomichi, T.; Kataoka, T. Betulinic acid and oleanolic acid, natural pentacyclic triterpenoids, interfere with N-linked glycan modifications to intercellular adhesion molecule-1, but not its intracellular transport to the cell surface. Eur. J. Pharmacol. 2015, 767, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Dye, D.E.; Coombe, D.R. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int. J. Cell Biol. 2012, 340296. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.M.; Salmi, C.; Loncle, C.; Vidal, N.; Letourneux, Y. Squalamine: A Polyvalent Drug of the Future? Curr. Cancer Drug Targets 2005, 5, 267–272. [Google Scholar] [CrossRef]
- Flekhter, O.B.; Nigmatullina, L.R.; Baltina, L.A.; Karachurina, L.T.; Galin, F.Z.; Zarudii, F.S.; Tolstikov, G.A.; Boreko, E.I.; Pavlova, N.I.; Nikolaeva, S.N.; et al. Synthesis of betulinic acid from betulin extract and study of the antiviral and antiulcer activity of some related terpenoids. Pharm. Chem. J. 2002, 36, 484–487. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Compounds | Mean Growth, % | Range of Growth, % | Sensitive Cell Lines, % |
|---|---|---|---|
| 2 | 89.47 | 8.04 to 126.62 | Leukemia: SR (8.04) |
| 7 | 99.22 | 53.80 to 146.79 | not active |
| 8 | 80.54 | 20.45 to 135.21 | Colon Cancer: HT29 (24.32); Breast Cancer: MDA-MB-231/ATCC (20.45) |
| 9 | 72.02 | 25.88 to 115.11 | Leukemia: HL-60(TB) (25.88) |
| 12 | 98.94 | 59.08 to 140.73 | not active |
| 13 | 104.76 | 85.83 to 118.03 | not active |
| 14 | 36.65 | ‒33.78 to 73.79 | Leukemia: CCRF-CEM (14.95), HL-60(TB) (−14.34), MOLT-4 (11.67), RPMI-8226 (22.20); Non-Small Cell Lung Cancer: HOP-92 (8.07), NCI-460 (20.60); Colon Cancer: HCT-116 (17.53), HT29 (23.68), SW-620 (28.83); CNS Cancer: SF-295 (31.97), SNB-75 (23.86), U251 (27.93); Melanoma: SK-MEL-5 (−8.44); Renal Cancer: ACHN (23.51), CAKI-1 (18.57), UO-31 (17.41); Prostate Cancer: PC-3 (19.12); Breast Cancer: MCF7 (22.21), MDA-MB-231/ATCC (−33.78), T-47D (28.02) |
| 15 | 90.11 | 56.79 to 114.03 | not active |
| 16 | 100.79 | 59.32 to 140.95 | not active |
| 17 | 98.74 | 79.25 to 111.36 | not active |
| 19 | 63.02 | −39.45 to 140.99 | Leukemia: CCRF-CEM (11.76), K-562 (1.52), MOLT-4 (10.06), RPMI-8226 (29.30), SR (18.41); Non-Small Cell Lung Cancer: NCI-460 (−16.23); Colon Cancer: COLO 205 (18.88), HCT-116 (12.60), HT29 (4.21), KM12 (31.48), SW-620 (7.33); CNS Cancer: U251 (−39.45); Melanoma: LOX IMVI (5.71); Ovarian cancer: IGROV1 (27.33), OVCAR-8 (−28.27); Breast Cancer: MCF7 (14.49) |
| 20 | 100.53 | 87.76 to 111.05 | not active |
| 22 | 101.76 | 80.54 to 123.09 | not active |
| 24 | 93.62 | 21.69 to 131.79 | Leukemia: SR (21.69) |
| 25 | 91.74 | 21.22 to 112.08 | Leukemia: SR (21.22) |
| 26 | 90.51 | 60.92 to 125.91 | not active |
| Subpanel Tumor Cell Lines | Percentage Cell Growth for Compounds | |||||
|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 10 | 11 | 21 | |
| Leukemia | ||||||
| CCRF-CEM | −12.36 | 3.11 | 5.18 | 1.39 | −11.29 | 7.42 |
| HL-60(TB) | −23.06 | 1.17 | −23.98 | 1.25 | −35.58 | 0.90 |
| K-562 | 3.10 | 4.57 | −15.78 | 1.10 | −16.99 | 5.42 |
| MOLT-4 | −29.06 | 1.86 | 1.18 | 2.76 | −16.31 | −26.81 |
| RPMI-8226 | −18.26 | −19.77 | −8.15 | −6.66 | −30.74 | 3.28 |
| SR | 10.81 | 15.50 | −4.72 | 23.66 | 4.96 | 1.86 |
| NSC Lung Cancer | ||||||
| A549/ATCC | 9.89 | −69.33 | −64.63 | −4.70 | −76.50 | 15.82 |
| EKVX | −25.21 | −10.31 | −11.36 | −15.68 | 11.37 | 53.13 |
| HOP-62 | −72.98 | −72.18 | −69.12 | −26.80 | 43.09 | 32.54 |
| HOP−92 | −83.15 | −62.83 | −65.35 | −55.49 | −65.19 | −14.95 |
| NCI-H226 | −58.51 | −50.47 | −23.56 | 36.64 | 74.30 | 2.42 |
| NCI-H23 | −40.35 | −27.08 | −51.70 | −8.47 | −35.23 | 21.79 |
| NCI-H322M | −78.19 | −58.91 | −83.84 | −68.83 | −81.08 | 46.12 |
| NCI-H460 | −61.81 | −37.45 | −100.00 | −43.19 | −90.93 | 4.23 |
| NCI-H522 | −61.13 | −53.76 | −57.83 | −16.98 | −69.70 | 13.36 |
| Colon Cancer | ||||||
| COLO 205 | −52.98 | −66.21 | −19.47 | 7.82 | −47.48 | 17.60 |
| HCC-2998 | −68.10 | −39.02 | −90.36 | −23.40 | −90.04 | 26.92 |
| HCT-116 | −84.18 | −24.80 | −26.53 | 1.97 | −80.70 | 4.16 |
| HCT-15 | −44.10 | 6.36 | −24.81 | −22.06 | −36.56 | 34.28 |
| HT29 | −77.68 | −49.47 | −39.56 | 3.90 | −77.42 | 3.88 |
| KM12 | −62.05 | −63.59 | −52.23 | −25.31 | −81.93 | 10.89 |
| SW-620 | −55.13 | −4.49 | −86.66 | −36.18 | −76.71 | 21.96 |
| CNS Cancer | ||||||
| SF-268 | −61.04 | 4.41 | −30.99 | −16.88 | −38.68 | 29.14 |
| SF-295 | −50.75 | −57.70 | −84.72 | −26.39 | −54.47 | 36.56 |
| SF-539 | −83.91 | −70.46 | −100.00 | −36.20 | −92.71 | −2.29 |
| SNB-19 | −39.95 | 1.30 | −74.52 | −29.49 | −78.54 | 28.93 |
| SNB-75 | −47.83 | −39.09 | −79.32 | −51.56 | −41.55 | 18.44 |
| U251 | −51.40 | −69.32 | −71.65 | −13.77 | −81.82 | −39.66 |
| Melanoma | ||||||
| LOX IMVI | −85.52 | −72.80 | −41.03 | 1.37 | −68.23 | 14.56 |
| MALME-3M | −88.97 | −57.05 | −100.00 | −18.76 | −72.82 | 25.27 |
| M14 | −44.25 | 8.80 | −30.98 | 5.07 | −62.82 | 26.57 |
| MDA-MB-435 | −63.78 | −4.44 | −89.21 | −32.41 | −49.89 | 14.55 |
| SK-MEL-2 | −77.63 | −31.18 | −65.39 | −24.21 | 56.36 | 36.23 |
| SK-MEL-28 | −73.19 | 0.38 | −98.68 | −31.42 | −73.91 | 29.25 |
| SK-MEL-5 | −71.94 | −39.59 | −90.10 | −23.93 | −86.18 | −84.68 |
| UACC-257 | −46.24 | −6.08 | −39.58 | −46.67 | −10.66 | 8.82 |
| UACC-62 | −82.97 | −23.23 | −100.00 | −22.62 | −77.94 | −25.76 |
| Ovarian Cancer | ||||||
| IGROV1 | −71.25 | −58.08 | −81.43 | −6.94 | −84.81 | 18.78 |
| OVCAR−3 | −76.19 | −46.59 | −66.02 | −45.89 | −72.48 | −34.55 |
| OVCAR-5 | −77.37 | −21.57 | −83.37 | −47.68 | −44.11 | 50.59 |
| OVCAR-8 | −31.70 | −64.82 | −12.90 | −3.81 | −75.03 | 13.17 |
| NCI/ADR-RES | −57.29 | −8.91 | −13.05 | −9.52 | −73.33 | 17.06 |
| SK-OV-3 | −11.89 | −45.72 | −14.17 | −27.22 | 70.49 | 34.60 |
| Renal Cancer | ||||||
| 786-0 | −73.99 | −72.16 | −83.51 | −22.61 | −84.46 | −11.47 |
| A498 | −81.70 | −28.33 | −32.64 | −44.82 | 80.81 | −0.38 |
| ACHN | −75.68 | −80.29 | −100.00 | −49.24 | −83.29 | 13.94 |
| CAKI-1 | −45.80 | −64.30 | −93.59 | −54.52 | −8.79 | 18.52 |
| RXF 393 | −64.94 | −49.35 | −82.88 | −42.28 | −71.88 | −43.14 |
| SN12C | −91.61 | −45.76 | −53.68 | −34.74 | −88.48 | −71.91 |
| TK-10 | −35.40 | −69.84 | −67.30 | −32.23 | −77.68 | 37.88 |
| UO-31 | −97.54 | −100.00 | −100.00 | −34.32 | −96.35 | 19.03 |
| Prostate Cancer | ||||||
| PC-3 | −47.42 | −24.47 | 1.07 | −53.78 | −24.64 | 4.49 |
| DU-145 | −69.87 | −32.96 | −8.34 | −100.00 | −56.67 | 27.53 |
| Breast Cancer | ||||||
| MCF7 | −45.58 | −12.52 | −60.54 | −8.46 | −57.97 | 12.44 |
| MDA-MB-231/ATCC | −88.58 | −28.40 | −56.94 | −47.78 | −92.38 | 5.09 |
| HS 578T | −32.99 | −1.70 | −23.26 | −4.98 | 47.76 | 24.26 |
| BT-549 | −55.13 | −21.60 | −51.94 | −2.14 | 58.31 | 4.40 |
| T-47D | −8.82 | −24.75 | −32.56 | −12.76 | −4.36 | 15.11 |
| MDA-MB-468 | −40.40 | −56.03 | −48.60 | −19.87 | −49.06 | 4.41 |
| Panel/Cell Line | 4 | 5 | 6 | 10 | 11 | 21 | DRB | 5-FU |
|---|---|---|---|---|---|---|---|---|
| GI50 a (µM) | GI50 (µM) | GI50 (µM) | GI50 (µM) | GI50 (µM) | GI50 (µM) | GI50 (µM) | GI50 (µM) | |
| Leukemia | ||||||||
| CCRF-CEM | 5.19 | 4.49 | 4.55 | 22.1 | 9.65 | 11.7 | 0.08 | 9.97 |
| HL-60(TB) | 2.76 | 3.59 | 7.33 | 16.5 | 8.88 | 5.18 | 0.19 | 2.30 |
| K-562 | 4.09 | 3.04 | 6.90 | 9.84 | 8.20 | 2.23 | - | 3.58 |
| MOLT-4 | 3.29 | 3.43 | 11.5 | 12.9 | 6.96 | 2.52 | 0.03 | 0.35 |
| RPMI-8226 | 2.63 | 3.53 | 2.81 | 17.2 | 6.78 | 4.91 | 0.08 | 0.04 |
| SR | 3.32 | 4.14 | 6.76 | 9.05 | 5.91 | 2.87 | 0.03 | - |
| Non-Small Cell Lung Cancer | ||||||||
| A549/ATCC | 3.97 | 2.11 | 4.97 | 19.6 | 7.69 | 5.27 | 0.06 | 0.18 |
| EKVX | 9.28 | 2.96 | 10.5 | 23.3 | 7.34 | 4.77 | 0.41 | - |
| HOP-62 | 1.10 | 3.21 | 16.5 | 18.3 | 7.91 | 12.5 | 0.07 | 0.39 |
| HOP-92 | 5.21 | 2.69 | 4.61 | 10.8 | 2.93 | 6.90 | 0.10 | 77.9 |
| NCI-H226 | 7.28 | 4.11 | 15.5 | 21.2 | 7.74 | 5.43 | 0.05 | 54.7 |
| NCI-H23 | 9.42 | 2.26 | 14.7 | 22.6 | 9.14 | 7.59 | 0.15 | 0.33 |
| NCI-H322M | 13.0 | 2.59 | 15.1 | 17.6 | 7.32 | >50.0 | - | - |
| NCI-H460 | 13.4 | 2.12 | 14.7 | 14.0 | 9.40 | 11.1 | 0.02 | 0.05 |
| NCI-H522 | 4.94 | 3.13 | 10.3 | 21.0 | 8.72 | 8.34 | 0.03 | 7.27 |
| Colon Cancer | ||||||||
| COLO 205 | 4.31 | 3.63 | 15.9 | 21.0 | 8.74 | 9.02 | 0.18 | 0.15 |
| HCC-2998 | 10.4 | 3.96 | 14.1 | 21.4 | 7.16 | 13.8 | 0.26 | 0.05 |
| HCT-116 | 3.57 | 1.97 | 4.40 | 13.9 | 6.65 | 2.89 | 0.08 | 0.22 |
| HCT-15 | 4.04 | 3.28 | 11.5 | 21.7 | 7.65 | 5.77 | 6.46 | 0.11 |
| HT29 | 3.73 | 2.56 | 14.6 | 6.95 | 7.17 | 6.70 | 0.12 | 0.17 |
| KM12 | 3.86 | 3.20 | 6.81 | 17.3 | 8.47 | 9.35 | 0.27 | 0.21 |
| SW-620 | 7.70 | 3.88 | 14.7 | 22.5 | 7.91 | 4.45 | 0.09 | 0.92 |
| CNS Cancer | ||||||||
| SF-268 | 6.95 | 4.34 | 13.5 | 23.0 | 7.53 | 11.4 | 0.10 | 1.62 |
| SF-295 | 8.57 | 3.39 | 13.8 | 22.6 | 9.42 | 21.4 | 0.10 | - |
| SF-539 | 5.52 | 1.78 | 11.1 | 17.9 | 7.53 | 11.0 | 0.12 | 0.06 |
| SNB-19 | 11.9 | 4.57 | 12.9 | 24.7 | 8.50 | 12.6 | 0.04 | 3.81 |
| SNB-75 | 5.59 | 2.74 | 11.1 | 32.1 | 6.26 | 31.2 | 0.07 | 78.7 |
| U251 | 5.10 | 2.54 | 12.7 | 17.7 | 7.34 | 4.30 | 0.04 | 0.92 |
| Melanoma | ||||||||
| LOX IMVI | 10.2 | 1.77 | 14.9 | 25.5 | 8.72 | 4.32 | 0.07 | 0.24 |
| MALME-3M | 10.5 | 2.39 | 14.6 | 23.3 | 7.40 | 11.8 | 0.12 | 0.05 |
| M14 | 7.38 | 6.05 | 11.0 | 24.7 | 8.38 | 11.9 | 0.18 | 0.98 |
| MDA-MB-435 | 5.23 | 8.65 | 13.4 | 28.7 | 8.70 | 9.70 | 0.25 | 0.07 |
| SK-MEL-2 | 10.8 | 7.63 | 16.8 | 27.3 | >50.0 | 13.8 | 0.17 | 56.7 |
| SK-MEL-28 | 12.0 | 1.09 | 16.0 | 22.1 | 8.49 | 17.1 | 0.21 | 1.03 |
| SK-MEL-5 | 3.80 | 5.22 | 11.1 | 19.4 | 6.98 | 3.02 | 0.08 | 0.46 |
| UACC-257 | 4.83 | 13.2 | 15.1 | 20.6 | 7.91 | 5.35 | 0.14 | 3.55 |
| UACC-62 | 5.46 | 4.20 | 9.29 | 27.4 | 8.96 | 2.32 | 0.12 | 0.52 |
| Ovarian Cancer | ||||||||
| IGROV1 | 13.2 | 3.32 | 16.1 | 21.5 | 6.42 | 14.9 | 0.17 | 1.22 |
| OVCAR-3 | 3.95 | 2.07 | 12.2 | 15.7 | 7.65 | 5.17 | 0.39 | 0.01 |
| OVCAR-4 | 4.72 | 3.54 | 14.1 | 25.6 | 7.39 | 12.3 | 0.37 | 4.43 |
| OVCAR-5 | 14.4 | 3.78 | 18.6 | 14.8 | 8.78 | 16.7 | 0.41 | 10.9 |
| OVCAR-8 | 54.4 | 3.12 | 18.8 | 27.7 | 8.41 | 9.66 | 0.10 | 1.74 |
| NCI/ADR-RES | 7.74 | 3.17 | 18.9 | 24.8 | 8.41 | 13.5 | 7.16 | 0.31 |
| SK-OV-3 | 13.4 | 8.18 | 17.5 | 17.7 | 10.6 | 10.8 | 0.22 | 21.8 |
| Renal Cancer | ||||||||
| 786-0 | 10.9 | 2.03 | 12.4 | 23.2 | 7.80 | 6.42 | 0.13 | 0.72 |
| A498 | 12.8 | 12.2 | 19.6 | 22.9 | 42.6 | 21.7 | 0.10 | 0.35 |
| ACHN | 5.78 | 2.44 | 12.0 | 21.7 | 13.8 | 10.5 | 0.08 | 0.27 |
| CAKI-1 | 3.24 | 1.98 | 5.92 | 21.9 | 5.87 | 10.2 | 0.95 | 0.07 |
| RXF 393 | 4.28 | 2.10 | 13.2 | 17.1 | 5.60 | 5.64 | 0.10 | 2.61 |
| SN12C | 5.65 | 3.00 | 13.5 | 20.8 | 7.56 | 7.28 | 0.07 | 0.49 |
| TK-10 | 14.7 | 2.79 | 15.8 | 23.9 | 10.1 | 13.8 | - | 1.12 |
| UO-31 | 7.57 | 1.72 | 11.7 | 38.9 | 6.85 | 15.6 | 0.49 | 1.42 |
| Prostate Cancer | ||||||||
| PC-3 | 2.67 | 2.83 | 2.95 | 12.6 | 6.26 | 2.26 | 0.32 | 2.36 |
| DU-145 | 9.94 | 3.21 | 14.1 | 17.5 | 7.47 | 13.4 | 0.11 | 0.36 |
| Breast Cancer | ||||||||
| MCF7 | 3.27 | 2.91 | 13.2 | 10.7 | 7.38 | 3.67 | 0.03 | 0.07 |
| MDA-MB-31/ATCC | 11.8 | 1.97 | 15.1 | 21.1 | 8.03 | 10.3 | 0.51 | 6.60 |
| HS 578T | 12.4 | 5.74 | 20.1 | 40.5 | 8.30 | 30.2 | 0.33 | 9.77 |
| BT-549 | 8.17 | 3.99 | 15.0 | 23.8 | 8.91 | 11.3 | 0.23 | 10.6 |
| T-47D | 2.91 | 4.55 | 10.9 | 13.1 | 7.52 | 3.47 | 0.06 | 8.12 |
| MDA-MB-468 | 3.96 | 3.20 | 8.88 | 12.6 | 8.09 | 2.02 | 0.05 | - |
| Panel | Compound 4 | Compound 5 | Compound 6 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MIDa | MIDb | Selectivityc | MIDa | MIDb | Selectivityc | MIDa | MIDb | Selectivityc | |
| Leukemia | 7.87 | 3.55 | 2.22 | 3.67 | 3.20 | 1.15 | 12.15 | 6.64 | 1.83 |
| NSCL cancer | 7.50 | 1.05 | 2.79 | 1.32 | 11.88 | 1.02 | |||
| Colon Cancer | 5.37 | 1.47 | 3.21 | 1.14 | 11.72 | 1.04 | |||
| CNS cancer | 7.27 | 1.08 | 3.23 | 1.14 | 12.52 | 0.97 | |||
| Melanoma | 7.80 | 1.01 | 5.58 | 0.66 | 13.58 | 0.89 | |||
| Ovarian Cancer | 15.97 | 0.49 | 3.88 | 0.95 | 14.10 | 0.86 | |||
| Renal Cancer | 8.12 | 0.97 | 3.53 | 1.04 | 13.02 | 0.93 | |||
| Prostate cancer | 6.31 | 1.25 | 3.02 | 1.22 | 8.53 | 1.42 | |||
| Breast cancer | 7.09 | 1.11 | 3.73 | 0.98 | 13.86 | 0.88 | |||
| Panel | Compound 10 | Compound 11 | Compound 21 | ||||||
| MIDa | MIDb | Selectivityc | MIDa | MIDb | Selectivityc | MIDa | MIDb | Selectivityc | |
| Leukemia | 20.46 | 14.59 | 1.40 | 9.17 | 7.73 | 1.19 | 10.25 | 4.90 | 2.09 |
| NSCL cancer | 18.71 | 1.09 | 7.58 | 1.21 | 12.43 | 0.83 | |||
| Colon Cancer | 17.82 | 1.15 | 7.68 | 1.19 | 7.42 | 1.38 | |||
| CNS cancer | 23.00 | 0.89 | 7.76 | 1.18 | 15.32 | 0.67 | |||
| Melanoma | 24.33 | 0.84 | 12.84 | 0.71 | 8.81 | 1.16 | |||
| Ovarian Cancer | 24.63 | 0.83 | 8.24 | 1.11 | 11.86 | 0.86 | |||
| Renal Cancer | 23.80 | 0.86 | 12.52 | 0.73 | 11.39 | 0.89 | |||
| Prostate cancer | 15.05 | 1.36 | 6.87 | 1.33 | 7.83 | 1.31 | |||
| Breast cancer | 20.30 | 1.01 | 8.04 | 1.14 | 10.16 | 1.00 | |||
| Compounds | Pearson’s Correlation b | P Value | NSC c | Name | Mechanism of Action | FDA Status |
|---|---|---|---|---|---|---|
| 4 | - | - | - | - | - | - |
| 5 | 0.545 | 0.000024 | 727038 | CDDO-Im | Nrf2 | - |
| 6 | 0.672 | 0 | 88536 | Calusterone | Hormone | FDA approved |
| 0.65 | 0 | 12198 | Dromostanolone propionate | Hormone | FDA approved | |
| 0.62 | 0 | 734945 | N-(4-Aminophenyl)-4-(3-(3,4-dihydroisoquinolin-2(1H)-yl)prop-1-en-2-yl)benzamide | HDAC | - | |
| 0.537 | 0.000012 | 736101 | 4-(3-(10H-Phenothiazin-10-yl)prop-1-en-2-yl)-N-(2-aminophenyl)benzamide | HDAC | - | |
| 0.565 | 0.000024 | 730001 | N-(2-Aminophenyl)-4-(3-(3,4-dihydro-1H-pyrido[3,4-b]indol-2(9H)-yl)prop-1-en-2-yl)benzamide | HDAC | - | |
| 0.507 | 0.000041 | 92339 | Fluphenazine | Antipsychotic | FDA approved | |
| 0.517 | 0.000063 | 734949 | 4-[3-(3,4-Dihydro-1H-isoquinolin-2-yl)prop-1-en-2-yl]-N-pyrazin-2-ylbenzamide | HDAC | - | |
| 10 | 0.563 | 0.000004 | 776422 | LDK-378 | ALK inhibitor | FDA approved |
| 0.537 | 0.000014 | 777193 | LDK-378 | ALK inhibitor | FDA approved | |
| 0.511 | 0.000035 | 12198 | Dromostanolone propionate | Hormone | FDA approved | |
| 11 | 0.507 | 0.000379 | 221019 | Wortmannin | PI3K inhibitor | Clinical trial |
| 21 | 0.578 | 0.000002 | 12198 | Dromostanolone propionate | Hormone | FDA approved |
| 0.537 | 0.000014 | 126771 | Dichloroallyl lawsone | DNA/RNA synthesis inhibitor | - |
| Compounds | Yeast α-Glucosidase, IC50 ± SE (μM) | Rat Liver ER Neutral α-Glucosidase Inhibition, m ± SD (%) | |
|---|---|---|---|
| 100 μM | 10 μM | ||
| 11 | 4.84 ± 1.02 | 43.60 ± 7.98 * | 15.03 ± 8.98 |
| 12 | >100 | 23.59 ± 1.34 * | 10.21 ± 3.49 |
| 16 | >100 | 14.94 ± 6.31 | 9.05 ± 6.68 |
| 18 | 5.70 ± 1.09 | 36.51 ± 14.90 * | 56.64 ± 4.24 * |
| 22 | >100 | 58.52 ± 8.21 * | 31.42 ± 7.96 * |
| 23 | >100 | 5.85 ± 8.03 | −1.34 ± 2.22 |
| 24 | >100 | 54.62 ± 15.38 * | 26.96 ± 2.33 * |
| 25 | 53.3 ± 5.7 | 9.62 ± 6.94 | 8.76 ± 9.63 |
| 26 | >100 | 26.60 ± 13.03 | 11.63 ± 9.09 |
| Acarbose | 436.7 ± 10.2 | 74.55 ± 3.76 * (1 mM) 24.09 ± 6.48 * (100 μM) | 5.68 ± 4.68 |
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | Fungi | ||||
|---|---|---|---|---|---|---|---|
| S. aureus | E. coli | K. pneumonia | P. aeruginosa | A. baumannii | C. albicans | C. neoformans | |
| Strain ATCC 43300 | Strain ATCC 25922 | Strain ATCC 700603 | Strain 19606 | Strain ATCC 27853 | Strain ATCC 90028 | Strain H99, ATCC 208821 | |
| 2 | 40.30 | 1.96 | 0.98 | 8.34 | 33.07 | 4.14 | −5.90 |
| 12 | 71.80 | −9.57 | 9.20 | 7.72 | 19.49 | 11.74 | 62.56 |
| 25 | 19.78 | −5.34 | 2.44 | −8.92 | 40.86 | 13.97 | −7.93 |
| 26 | 12.66 | −7.03 | −3.89 | −4.64 | 28.24 | 5.83 | −13.04 |
Simple Availability: Simples of the compounds 1–26 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazakova, O.B.; Giniyatullina, G.V.; Mustafin, A.G.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A. Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids. Molecules 2020, 25, 4833. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204833
Kazakova OB, Giniyatullina GV, Mustafin AG, Babkov DA, Sokolova EV, Spasov AA. Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids. Molecules. 2020; 25(20):4833. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204833
Chicago/Turabian StyleKazakova, Oxana B., Gul’nara V. Giniyatullina, Akhat G. Mustafin, Denis A. Babkov, Elena V. Sokolova, and Alexander A. Spasov. 2020. "Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids" Molecules 25, no. 20: 4833. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204833