
Discovery of Novel Dual Extracellular Regulated Protein Kinases (ERK) and Phosphoinositide 3-Kinase (PI3K) Inhibitors as a Promising Strategy for Cancer Therapy

 ,
,
Abstract
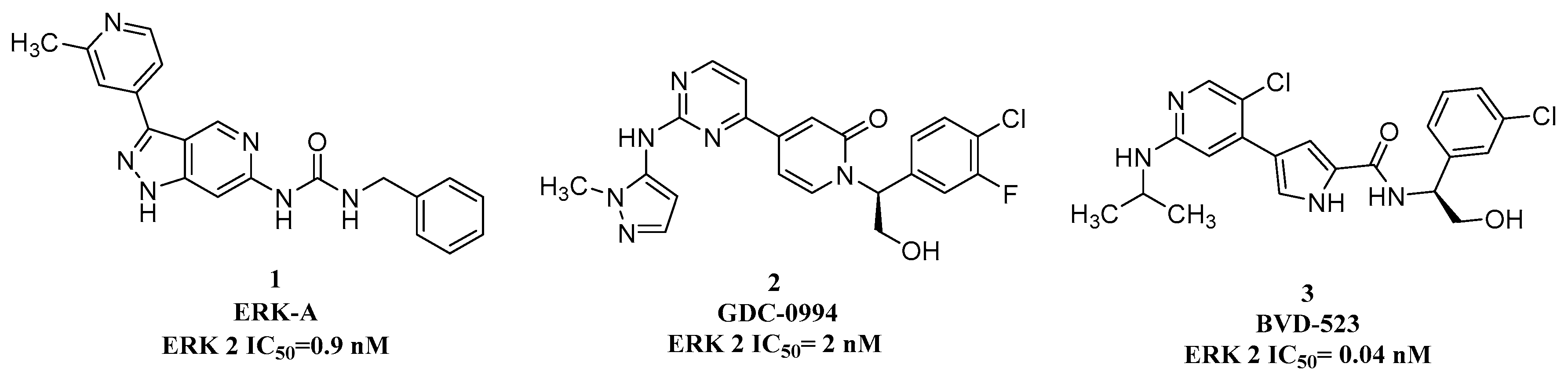
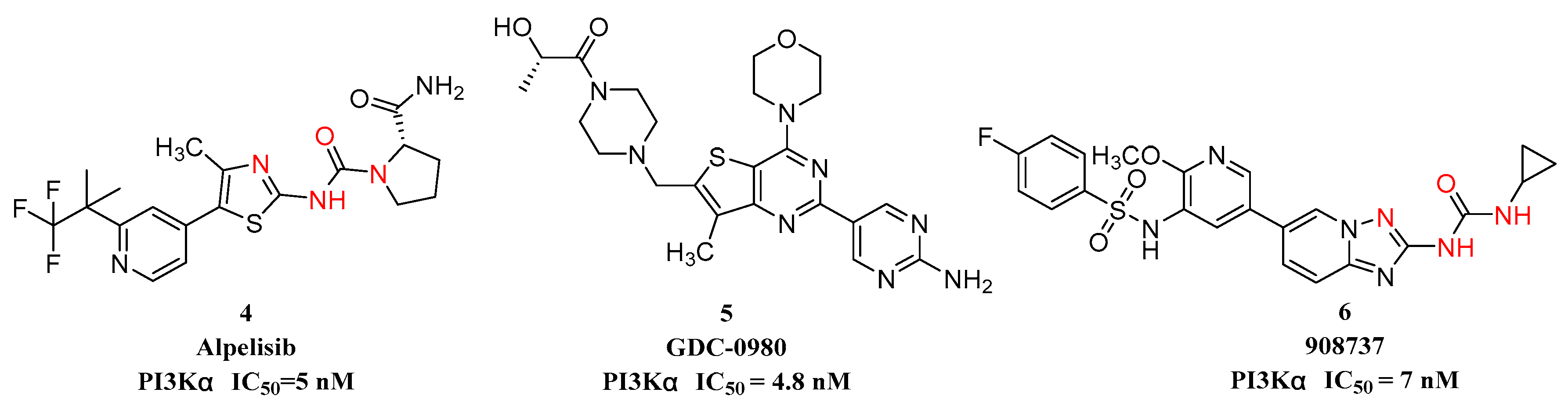
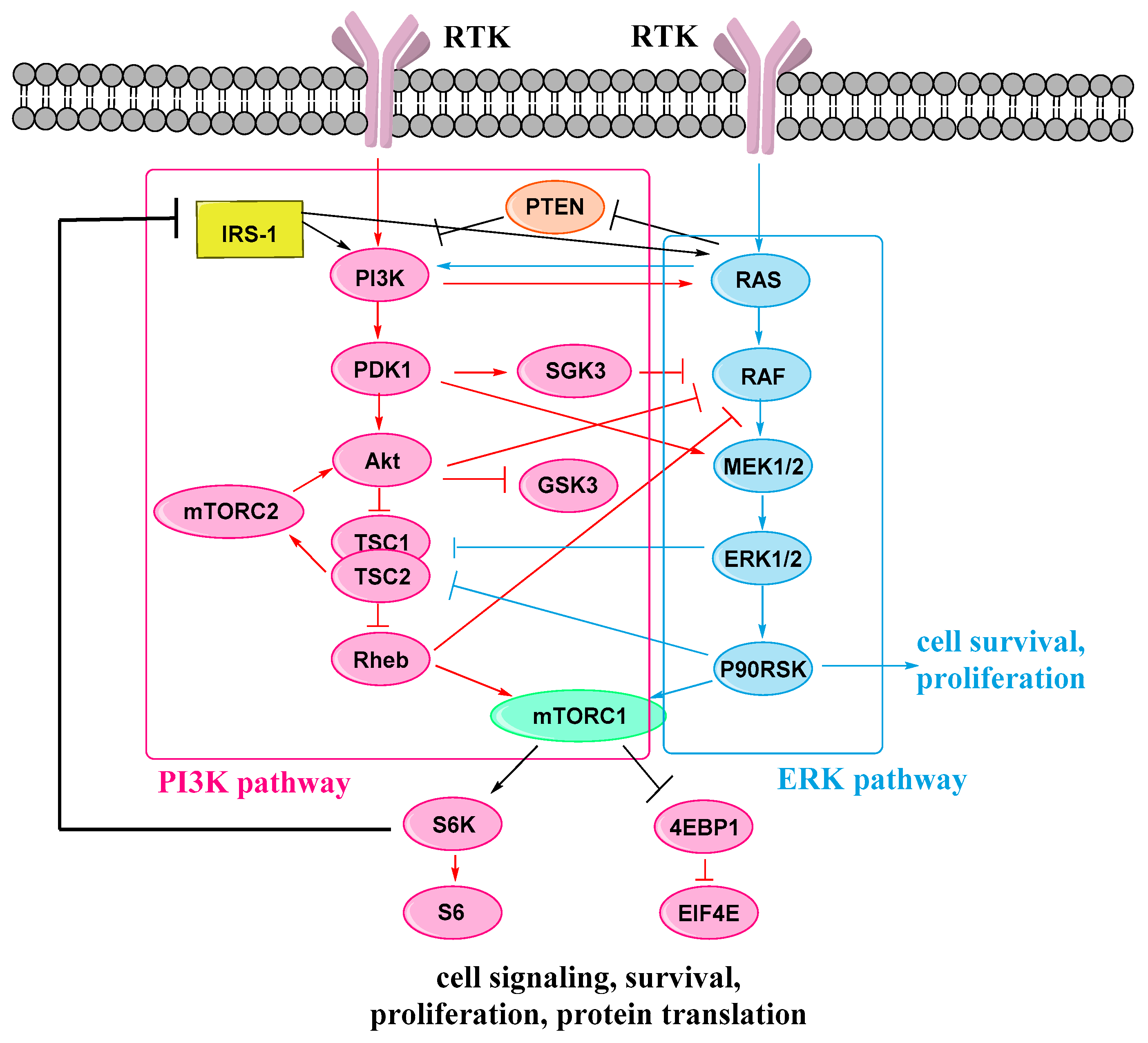
:1. Introduction
2. Results and Discussion
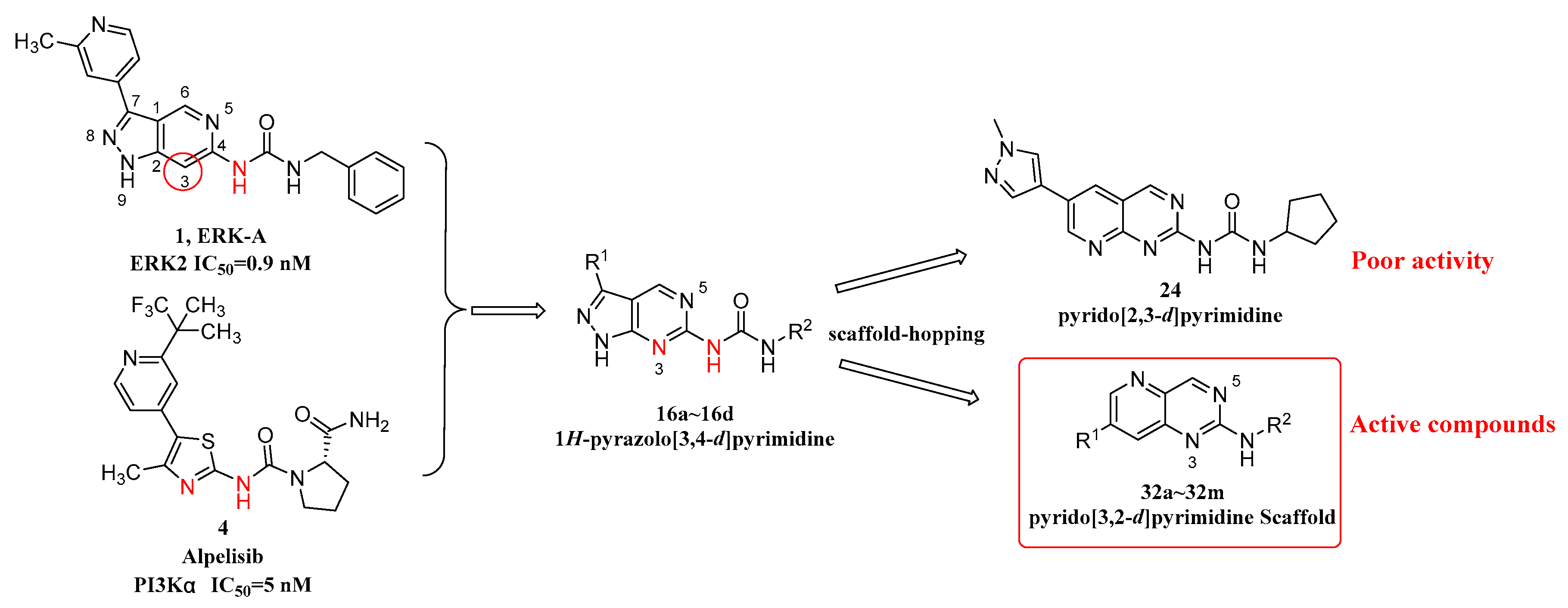
2.1. Drug Design
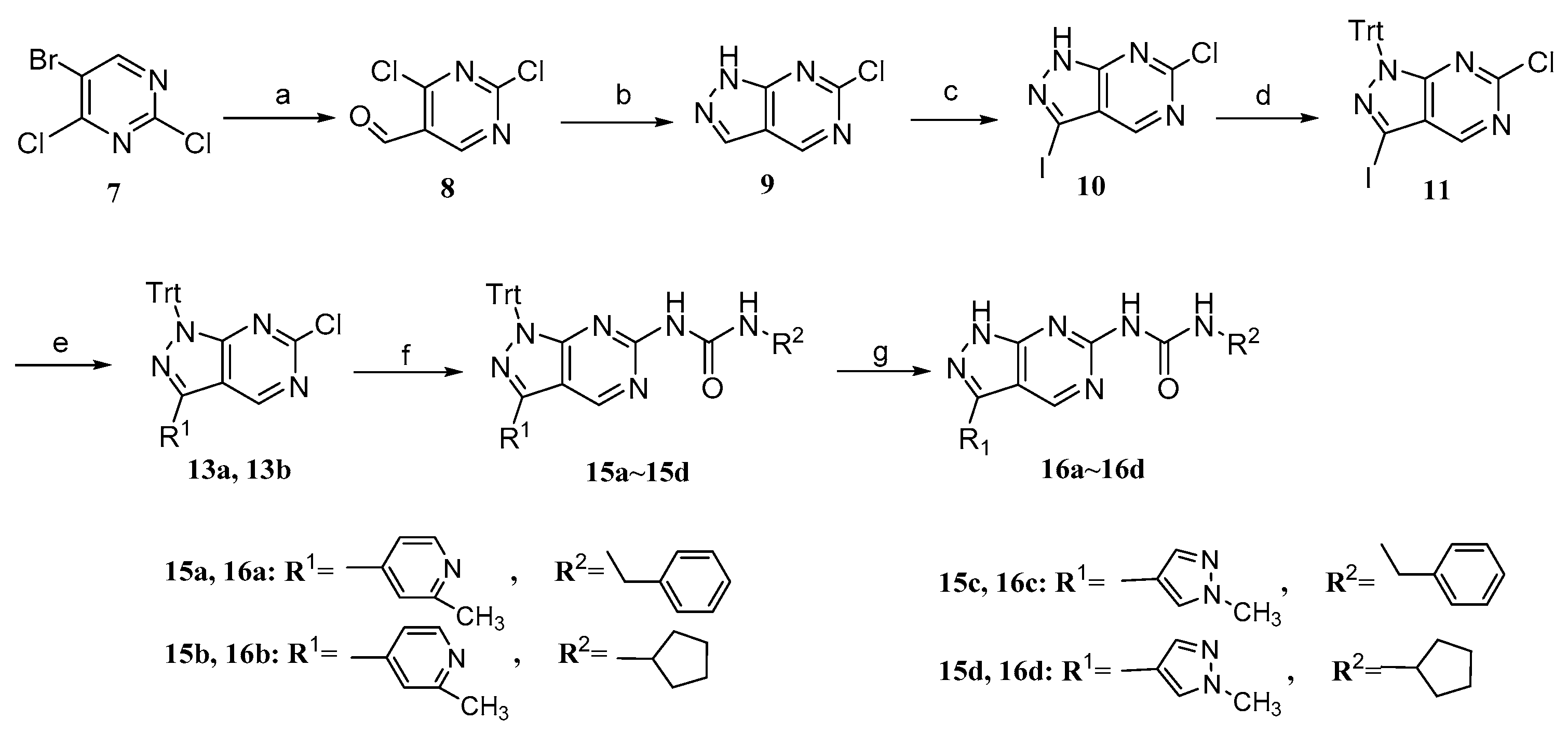
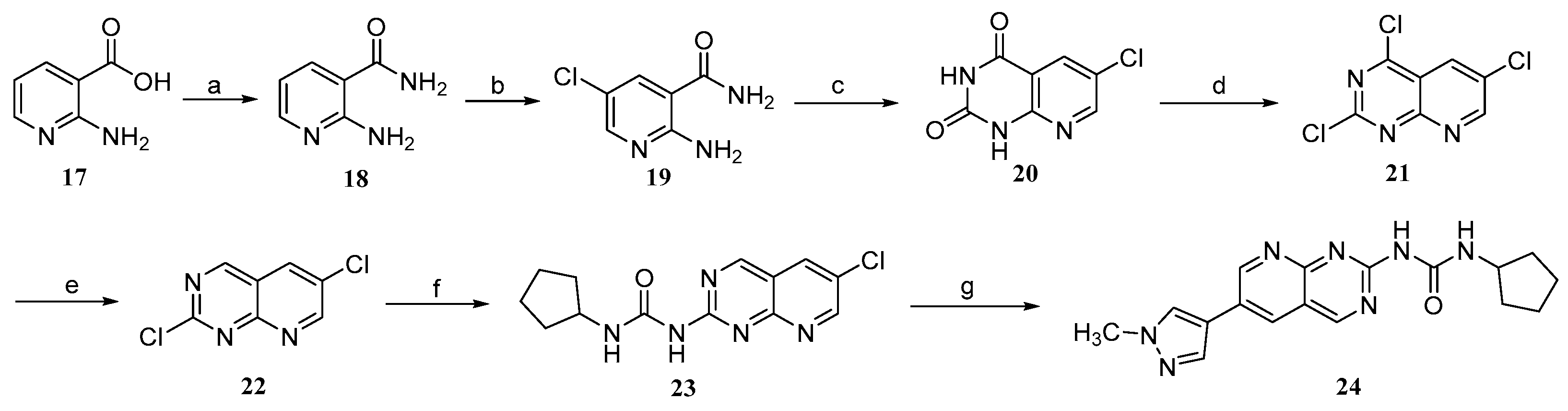
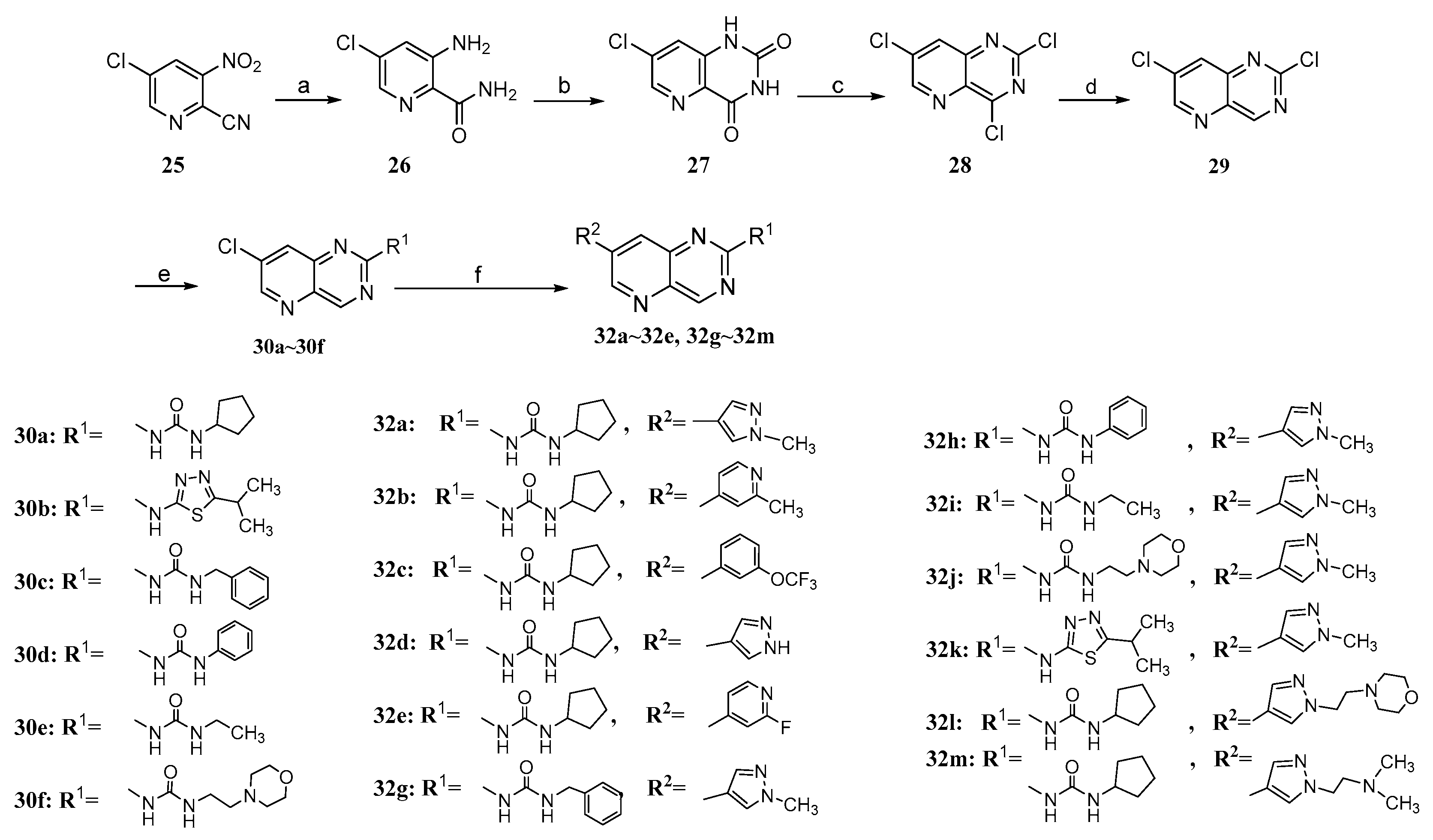
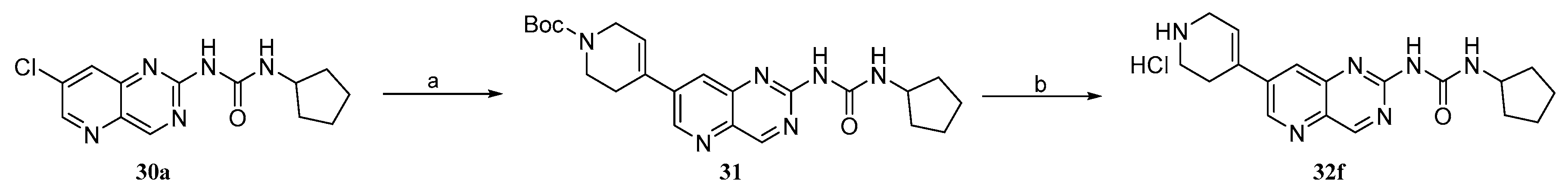
2.2. Chemistry
2.3. In Vitro ERK and PI3K Inhibition Assay
2.4. In Vitro Anti-Proliferation Assay
2.5. In Vitro Pharmacokinetic (PK) Profile
2.6. In Vivo Pharmacokinetic (PK) Profile
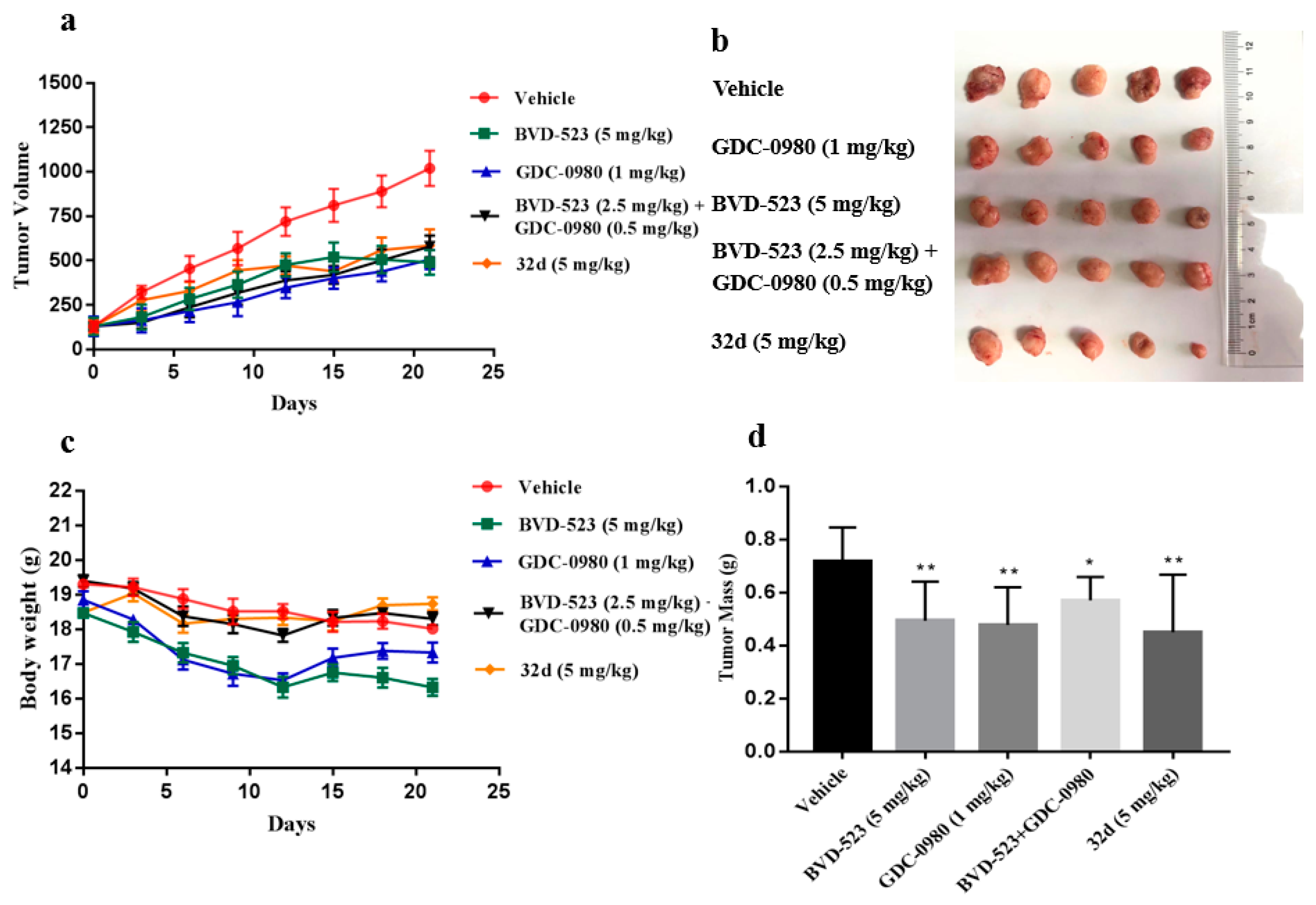
2.7. In Vivo Antitumor Activity Evaluation
3. Materials and Methods
3.1. Synthesis
3.1.1. 2,4-Dichloropyrimidine-5-carbaldehyde (8)
3.1.2. 6-Chloro-1H-pyrazolo[3,4-d]pyrimidine (9)
3.1.3. 6-Chloro-3-iodo-1H-pyrazolo[3,4-d]pyrimidine (10)
3.1.4. 6-Chloro-3-iodo-1-trityl-1H-pyrazolo[3,4-d]pyrimidine (11)
3.1.5. General Procedure for the Synthesis of 13a, 13b
6-Chloro-3-(2-methylpyridin-4-yl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidine (13a)
6-Chloro-3-(1-methyl-1H-pyrazol-4-yl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidine (13b)
3.1.6. General Procedure for the Synthesis of 15a~15d
1-Benzyl-3-(3-(2-methylpyridin-4-yl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (15a)
1-Cyclopentyl-3-(3-(2-methylpyridin-4-yl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (15b)
1-Benzyl-3-(3-(1-methyl-1H-pyrazol-4-yl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (15c)
1-Cyclopentyl-3-(3-(1-methyl-1H-pyrazol-4-yl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (15d)
3.1.7. 2-Aminonicotinamide (18)
3.1.8. 2-Amino-5-chloronicotinamide (19)
3.1.9. 6-Chloropyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (20)
3.1.10. 2,4,6-Trichloropyrido[2,3-d]pyrimidine (21)
3.1.11. 2,6-Dichloropyrido[2,3-d]pyrimidine (22)
3.1.12. 1-(6-Chloropyrido[2,3-d]pyrimidin-2-yl)-3-cyclopentylurea (23)
3.1.13. 3-Amino-5-chloropicolinamide (26)
3.1.14. 7-Chloropyrido[3,2-d]pyrimidine-2,4(1H,3H)-dione (27)
3.1.15. 2,4,7-Trichloropyrido[3,2-d]pyrimidine (28)
3.1.16. 2,7-Dichloropyrido[3,2-d]pyrimidine (29)
3.1.17. General Procedure for the Synthesis of 30a~30f
1-(7-Chloropyrido[3,2-d]pyrimidin-2-yl)-3-cyclopentylurea (30a)
N-(7-chloropyrido[3,2-d]pyrimidin-2-yl)-5-methyl-1,3,4-thiadiazol-2-amine (30b)
1-Benzyl-3-(7-chloropyrido[3,2-d]pyrimidin-2-yl)urea (30c)
1-(7-Chloropyrido[3,2-d]pyrimidin-2-yl)-3-phenylurea (30d)
1-(7-Chloropyrido[3,2-d]pyrimidin-2-yl)-3-ethylurea (30e)
1-(7-Chloropyrido[3,2-d]pyrimidin-2-yl)-3-(2-morpholinoethyl)urea (30f)
3.1.18. General Procedure for the Synthesis of 16a~16d
1-Benzyl-3-(3-(2-methylpyridin-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (16a)
1-Cyclopentyl-3-(3-(2-methylpyridin-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (16b)
1-Benzyl-3-(3-(1-methyl-1H-pyrazol-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (16c)
1-Cyclopentyl-3-(3-(1-methyl-1H-pyrazol-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl)urea (16d)
3.1.19. General Procedure for the Synthesis of 24, 32a~32e and 32g~32m
1-Cyclopentyl-3-(6-(1-methyl-1H-pyrazol-4-yl)pyrido[2,3-d]pyrimidin-2-yl)urea (24)
1-Cyclopentyl-3-(6-(1-methyl-1H-pyrazol-4-yl)pyrido[2,3-d]pyrimidin-2-yl)urea (32a)
1-Cyclopentyl-3-(7-(2-methylpyridin-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea (32b)
1-Cyclopentyl-3-(7-(3-(trifluoromethoxy)phenyl)pyrido[3,2-d]pyrimidin-2-yl)urea (32c)
1-(7-(1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl)-3-cyclopentylurea (32d)
1-Cyclopentyl-3-(7-(2-fluoropyridin-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea (32e)
1-Benzyl-3-(7-(1-methyl-1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea (32g)
1-(7-(1-Methyl-1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl)-3-phenylurea (32h)
1-Ethyl-3-(7-(1-methyl-1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea (32i)
1-(7-(1-Methyl-1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl)-3-(2-morpholinoethyl)urea (32j)
1-Cyclopentyl-3-(7-(2-methylpyridin-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea (32k)
1-Cyclopentyl-3-(7-(1-(2-morpholinoethyl)-1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea (32l)
1-Cyclopentyl-3-(7-(1-(2-(dimethylamino)ethyl)-1H-pyrazol-4-yl)pyrido[3,2-d]pyrimidin-2-yl) urea (32m)
3.1.20. tert-Butyl 4-(2-(3-cyclopentylureido)pyrido[3,2-d]pyrimidin-7-yl)-3,6-dihydropyridine-1(2H)-carboxylate (31)
3.1.21. 1-Cyclopentyl-3-(7-(1,2,3,6-tetrahydropyridin-4-yl)pyrido[3,2-d]pyrimidin-2-yl)urea hydrochloride (32f)
3.2. Enzymatic Inhibition Assays
3.3. Anti-Proliferation Assay
3.4. In Vitro Pharmacokinetic Study
3.5. In Vivo Pharmacokinetic Study
3.6. In Vivo Antitumor Activity Evalution
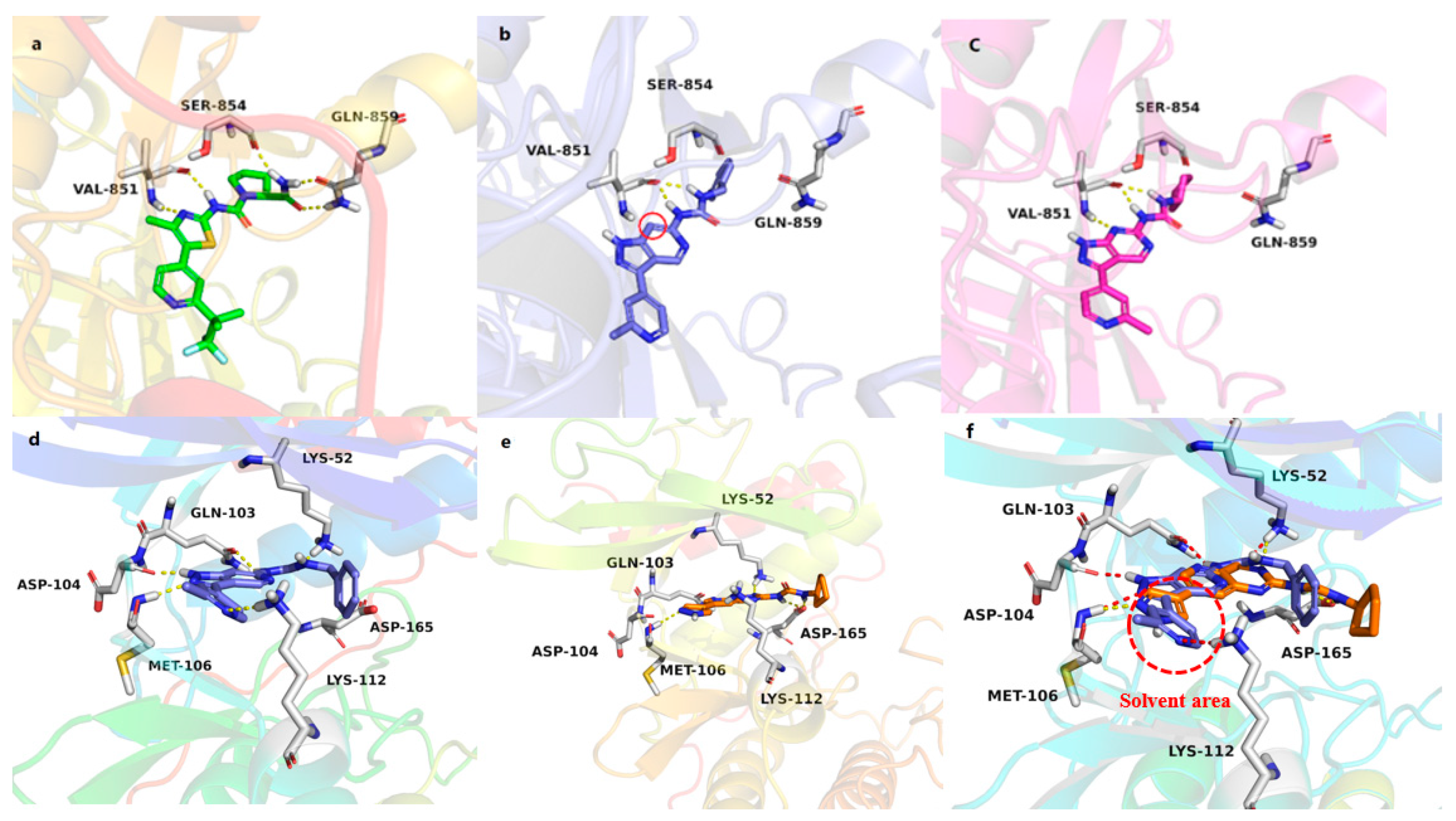
3.7. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pines, G.; Fankhauser, R.G.; Eckert, C.A. Predicting drug resistance using deep mutational scanning. Molecules 2020, 25, 2265. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Bardia, A.; Gounder, M.M.; Rodon, J.; Janku, F.; Lolkema, M.P.; Stephenson, J.J.; Bedard, P.L.; Schuler, M.; Sessa, C.; Lorusso, P.; et al. Phase Ib study of combination therapy with MEK inhibitor binimetinib and phosphatidylinositol 3-kinase inhibitor buparlisib in patients with advanced solid tumors with RAS/RAF alterations. Oncologist 2020, 25, e160–e169. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Robbins, D.J.; Zhen, E.; Owaki, H.; Vanderbilt, C.A.; Ebert, D.; Geppert, T.D.; Cobb, M.H. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 1993, 268, 5097–5106. [Google Scholar]
- Hancock, C.N.; Macias, A.; Lee, E.K.; Yu, S.Y.; MacKerell, A.D.; Shapiro, P. Identifcation of novel extracellular signal-regulated kinase docking domain inhibitors. J. Med. Chem. 2005, 48, 4586–4595. [Google Scholar] [CrossRef]
- Sammons, R.M.; Ghose, R.; Tsai, K.; Dalby, K.N. Targeting ERK beyond the boundaries of the kinase active site in melanoma. Mol. Carcinog. 2019, 58, 1551–1570. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Kelley, E.H.; Methot, J.L.; Zhou, H.; Petrocchi, A.; Chen, H.; Hill, S.E.; Hinton, M.C.; Hruza, A.; Jung, J.O.; et al. Discovery of 1-(1H-Pyrazolo[4,3-c]pyridin-6-yl)urea inhibitors of extracellular signal-regulated kinase (ERK) for the treatment of cancers. J. Med. Chem. 2016, 59, 6501–6511. [Google Scholar] [CrossRef]
- Blake, J.F.; Burkard, M.; Chan, J.; Chen, H.; Chou, K.J.; Diaz, D.; Dudley, D.A.; Gaudino, J.J.; Gould, S.E.; Grina, J.; et al. Discovery of (S) 1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl 1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H) one (GDC-0994), an extracellular signal-regulated kinase 1/2 (ERK1/2) inhibitor in early clinical development. J. Med. Chem. 2016, 59, 5650–5660. [Google Scholar] [CrossRef] [Green Version]
- James, M.C.; Geoffrey, I.S. Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr. Oncol. Rep. 2010, 12, 87–94. [Google Scholar]
- Pulido, R. PTEN inhibition in human disease therapy. Molecules 2018, 23, 285. [Google Scholar] [CrossRef] [Green Version]
- Sathe, A.; Chalaud, G.; Oppolzer, I.; Wong, K.Y.; Von Busch, M.; Schmid, S.C.; Tong, Z.; Retz, M.; Gschwend, J.E.; Schulz, W.A.; et al. Parallel PI3K, AKT and mTOR inhibition is required to control feedback loops that limit tumor therapy. Molecules 2018, 13, e0190854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Sutherlin, D.P.; Bao, L.; Berry, M.; Castanedo, G.; Chuckowree, I.; Dotson, J.; Folks, A.; Friedman, L.; Goldsmith, R.; Gunzner, J.; et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J. Med. Chem. 2011, 54, 7579–7587. [Google Scholar] [CrossRef]
- Wang, X.M.; Mao, S.; Cao, L.; Xie, X.X.; Xin, M.H.; Lian, J.F.; Cao, Y.; Zhang, S. Modification of N-(6-(2-methoxy-3-(4-fluorophenyl sulfonamido) pyridin-5-yl)-[1,2,4] triazolo [1,5-a] pyridin-2-yl) acetamide as PI3Ks inhibitor by replacement of the acetamide group with alkylurea. Bioorganic Med. Chem. 2015, 23, 5662–5671. [Google Scholar] [CrossRef]
- Won, J.K.; Yang, H.W.; Shin, S.Y.; Lee, J.H.; Heo, W.D.; Cho, K.H. The cross regulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J. Mol. Cell Biol. 2012, 4, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Rambur, A.; Lours-Calet, C.; Beaudoin, C.; Buñay, J.; Vialat, M.; Mirouse, V.; Trousson, A.; Renaud, Y.; Lobaccaro, J.-M.; Baron, S.; et al. Sequential Ras/MAPK and PI3K/AKT/mTOR pathways recruitment drives basal extrusion in the prostate-like gland of Drosophila. Nat. Commun. 2020, 11, 2300. [Google Scholar] [CrossRef]
- Dalezis, P.; Geromichalou, E.; Polonifi, A.; Sagredou, S.; Nikoleousakos, N.; Nikolaou, M.; Sarli, V.; Panayiotidis, M.I.; Trafalis, D. Azasteroid Alkylators as dual inhibitors of AKT and ERK signaling for the treatment of ovarian carcinoma. Cancers 2020, 12, 1263. [Google Scholar] [CrossRef]
- Heard, J.J.; Phung, I.; Potes, M.I.; Tamanoi, F. An oncogenic mutant of RHEB, RHEB Y35N, exhibits an altered interaction with BRAF resulting in cancer transformation. BMC Cancer 2018, 18, 69. [Google Scholar] [CrossRef] [Green Version]
- Chiang, Y.J.; Liao, W.T.; Ho, K.C.; Wang, S.H.; Chen, Y.G.; Ho, C.L.; Huang, S.; Shih, L.; Yang-Yen, H.; Yen, J.J. CBAP modulates Akt-dependent TSC2 phosphorylation to promote Rheb-mTORC1 signaling and growth of T-cell acute lymphoblastic leukemia. Oncogene 2019, 38, 1432–1447. [Google Scholar] [CrossRef]
- Saini, K.S.; Loi, S.; De Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.J.; Piccart-Gebhart, M. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R1 | R2 | ERK2 Inhibition% (1 μM) | PI3Kα Inhibition% (1 μM) |
|---|---|---|---|---|
| 16a |  |  | 98.3 | 8.7 |
| 16b |  |  | 84.8 | 27.4 |
| 16c |  |  | 97.2 | 1.8 |
| 16d |  |  | 79.5 | 4.0 |
| BVD-525 | 97.7 | ND | ||
| BYL-719 | ND | 96.7 |

| Entry | AB | ERK2 Inhibition% (1 μM) | PI3Kα Inhibition% (1 μM) |
|---|---|---|---|
| 24 |  | 1.8 | 15.1 |
| 32a |  | 89.9 | 85.0 |

| Entry | R1 | R2 | ERK2 Inhibition% (100 nM) | PI3Kα Inhibition% (100 nM) |
|---|---|---|---|---|
| 32b |  |  | 53.7 | 10.5 |
| 32c |  |  | 1.6 | −11.3 |
| 32d |  |  | 49.3 | 51.4 |
| 32e |  |  | 35.4 | 7.0 |
| 32f |  |  | 0.4 | −8.8 |
| 32g |  |  | 55.2 | 32.4 |
| 32h |  |  | 17.2 | 24.9 |
| 32i |  |  | 22.1 | 33.4 |
| 32j |  |  | 6.2 | 15.1 |
| 32k |  |  | 66.5 | 15.3 |
| 32l |  |  | 21.4 | 63.8 |
| 32m |  |  | 8.4 | 41.0 |
| BVD-525 | 97.7 | ND | ||
| BYL-719 | ND | 98.2 |

| Entry | R1 | R2 | ERK2 IC50 (nM) | PI3Kα IC50 (nM) |
|---|---|---|---|---|
| 32a |  |  | 89 | 38 |
| 32d |  |  | 73 | 59 |
| 32g |  |  | 83 | 824 |
| Entry | ERK1 Inhibition% (100 nM) | PI3Kβ Inhibition% (100 nM) | PI3Kγ Inhibition% (100 nM) | PI3Kδ Inhibition% (100 nM) |
|---|---|---|---|---|
| 32a | 39.3% | 13.9% | 43.2% | 11.1% |
| Entry | IC50 (μM) | |
|---|---|---|
| HEC1B | HCT116 | |
| 32a | 1.848 | 1.945 |
| 32d | 1.492 | 0.7255 |
| 32g | 2.272 | 10 |
| 32l | 3.63 | 1.76 |
| BVD-523 | 18.02 | 0.3209 |
| GDC-0980 | 0.2295 | 1.065 |
| Parameters | Value |
|---|---|
| K (min−1) | 0.004 |
| t1/2 (min) | 173.25 |
| Pm (mg/mL) | 0.25 |
| Clint (mL/min/mg) | 0.016 |
| Comp. i.v./p.o. | Dose mg/kg | CL mL/kg/ min | Vz mL/kg | AUC0–t ng·h/mL | AUC0–∞ ng·h/mL | MRT0–t h | t1/2 h | Cmax ng/mL | F% |
|---|---|---|---|---|---|---|---|---|---|
| 32d (i.v.) | 1 | 44.8 | 9124 | 368.0 | 379.0 | 0.99 | 2.32 | 1306.7 | -- |
| 32d (p.o.) | 10 | 727.9 | 244740 | 191.5 | 355.1 | 2.93 | 2.65 | 128.8 | 9.37 |
Sample Availability: Samples of the compounds are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ju, Q.; Sun, J.; Huang, L.; Wu, S.; Wang, S.; Li, Y.; Guan, Z.; Zhu, Q.; Xu, Y. Discovery of Novel Dual Extracellular Regulated Protein Kinases (ERK) and Phosphoinositide 3-Kinase (PI3K) Inhibitors as a Promising Strategy for Cancer Therapy. Molecules 2020, 25, 5693. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235693
Zhang L, Ju Q, Sun J, Huang L, Wu S, Wang S, Li Y, Guan Z, Zhu Q, Xu Y. Discovery of Novel Dual Extracellular Regulated Protein Kinases (ERK) and Phosphoinositide 3-Kinase (PI3K) Inhibitors as a Promising Strategy for Cancer Therapy. Molecules. 2020; 25(23):5693. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235693
Chicago/Turabian StyleZhang, Lingzhi, Qiurong Ju, Jinjin Sun, Lei Huang, Shiqi Wu, Shuping Wang, Yin Li, Zhe Guan, Qihua Zhu, and Yungen Xu. 2020. "Discovery of Novel Dual Extracellular Regulated Protein Kinases (ERK) and Phosphoinositide 3-Kinase (PI3K) Inhibitors as a Promising Strategy for Cancer Therapy" Molecules 25, no. 23: 5693. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235693