
Myricetin as a Promising Molecule for the Treatment of Post-Ischemic Brain Neurodegeneration


1
Laboratory of Ischemic and Neurodegenerative Brain Research, Mossakowski Medical Research Institute, Polish Academy of Sciences, 02-106 Warsaw, Poland
2
Department of Pathophysiology, Medical University of Lublin, 20-090 Lublin, Poland
*
Author to whom correspondence should be addressed.
Nutrients 2021, 13(2), 342; https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020342
Submission received: 15 December 2020
/
Revised: 17 January 2021
/
Accepted: 20 January 2021
/
Published: 24 January 2021
(This article belongs to the Special Issue Bioactive Natural Compounds for Therapeutic and Nutraceutical Applications in Neurodegeneration)
{kind=link}
{kind=link}
Abstract
:The available drug therapy for post-ischemic neurodegeneration of the brain is symptomatic. This review provides an evaluation of possible dietary therapy for post-ischemic neurodegeneration with myricetin. The purpose of this review was to provide a comprehensive overview of what scientists have done regarding the benefits of myricetin in post-ischemic neurodegeneration. The data in this article contribute to a better understanding of the potential benefits of myricetin in the treatment of post-ischemic brain neurodegeneration, and inform physicians, scientists and patients, as well as their caregivers, about treatment options. Due to the pleiotropic properties of myricetin, including anti-amyloid, anti-phosphorylation of tau protein, anti-inflammatory, anti-oxidant and autophagous, as well as increasing acetylcholine, myricetin is a promising candidate for treatment after ischemia brain neurodegeneration with full-blown dementia. In this way, it may gain interest as a potential substance for the prophylaxis of the development of post-ischemic brain neurodegeneration. It is a safe substance, commercially available, inexpensive and registered as a pro-health product in the US and Europe. Taken together, the evidence available in the review on the therapeutic potential of myricetin provides helpful insight into the potential clinical utility of myricetin in treating neurodegenerative disorders with full-blown dementia. Therefore, myricetin may be a promising complementary agent in the future against the development of post-ischemic brain neurodegeneration. Indeed, there is a scientific rationale for the use of myricetin in the prevention and treatment of brain neurodegeneration caused by ischemia.
1. Introduction
With the aging of the world population, ischemic stroke has become the second leading cause of death in people aged 60 and over, and the fifth leading cause of death in people aged 15 to 59 worldwide [1]. Worldwide, 70% cases of ischemic stroke and 87% of deaths related to ischemic stroke and disability adjusted life years occur in low- and middle-income countries [2]. The incidence of ischemic stroke cases has more than doubled in low- and middle-income countries in the last four decades [2]. Over these decades, the incidence of ischemic stroke cases has decreased by 42% in high-income countries [2]. Ischemic stroke occurs on average 15 years earlier in people living in low and middle-income countries and causes more deaths compared to those in high-income countries [3]. As many as 84% of ischemic stroke patients in low- and middle-income countries die within three years of stroke, compared with 16% in high-income countries [2]. Current epidemiological statistics indicate that approximately 17 million people suffer from ischemic brain injury annually, of whom 6 million die each year [4,5]. Worldwide, the number of post-ischemia cases has now reached approximately 33 million [4,5]. According to the latest forecasts, the number of cases will increase in the future to 77 million in 2030 [4,5]. In 2010, the annual cost of treating stroke patients in Europe was approximately € 64 billion [5]. If the trend in ischemic stroke continues, there will be approximately 12 million deaths by 2030, 70 million will be stroke survivors, and more than 200 million disability-adjusted life years will be recorded worldwide annually [5].
The mechanisms of post-ischemic neurodegeneration are complex and unclear, and the development of it is influenced by many factors. Excessive amyloid accumulation and increased tau protein hyperphosphorylation are currently the most studied factors in post-ischemic neurodegeneration with dementia [6,7,8]. It is also believed that metabolic imbalances in the brain, such as hyperactivity of the glutamate system, acetylcholine deficiency and metal ion dyshomeostasis, are closely related to the development of neurodegeneration as a consequence of ischemic brain injury [9,10,11,12]. Moreover, abnormal processes such as oxidative stress, neuroinflammation, and impaired autophagy have been found to cause severe brain damage and contribute to progressive and irreversible damage following reversible ischemia [13,14,15,16,17,18,19]. Dyshomeostasis of the intestinal flora is also mentioned as the driving force in the genesis and development of neurodegeneration after ischemia [20,21].
In the treatment of stroke, the first priority is to focus on physical recovery, but in the first year after stroke, 4 out of 10 patients have some degree of cognitive impairment without dementia [22]. This may be related to demographic and disease factors. About 6 months after ischemic stroke, women with a history of cerebrovascular disease and patients with lacunar infarction develop new cognitive impairment [23]. Identification of dementia immediately after stroke is difficult because of additional persistent deficits after ischemic stroke, both in terms of global cognition and individual domains, e.g., attention and processing speed, memory, language, and frontal executive functions may be impaired [24]. Finally, it is true that a history of ischemic stroke is a strong independent risk factor for the development of dementia [8,24,25,26,27,28]. Brain ischemia accelerates the onset of dementia by 10 years [29], and 10% of people develop dementia soon after the first incidence of ischemic stroke and 41.3% after the recurrent stroke [24,30].
A transient episode of cerebral ischemia in humans and animals causes acute massive neuronal loss in the CA1 region of the hippocampus and the cerebral cortex [6,7,31,32,33,34,35,36]. In these structures, necrotic and apoptotic neurons were mixed with neurons damaged after ischemia [6,31,32,35,36]. After a short-term post-ischemic survival, the number of damaged neurons decreased while the number of dead neurons increased [31,32]. After prolongation of post-ischemia survival, acute and chronic neuronal changes were observed in ischemia-resistant areas, in addition to acute neuronal death in ischemia-sensitive areas like hippocampal CA1 subfield [31,32,33,34]. The lesions were present in areas of the brain not affected by the primary ischemic injury, such as the CA2, CA3, and CA4 areas of the hippocampus [31,32,33,34,35,36]. Following ischemia, neuronal loss was observed with a decrease in acetylcholine levels in the hippocampus, suggesting that an additional cause of neuronal death was a deficiency in neuronal excitation [9,12,37,38]. In addition, ultrastructural studies revealed changes in hippocampal synapses after ischemia [38,39]. Other observations showed that an episode of brain ischemia with recirculation led to the induction of synaptic autophagy, which may be associated with the death of pyramidal neurons in the hippocampus after transient reversible cerebral ischemia [9,16,17,18,37,38,39,40,41,42].
Alterations in white matter and activation of glial cells in the brain tissue have been reported in both humans and animals following ischemia with recirculation [13,19,31,32,43,44,45]. In animal models of reversible cerebral ischemia, an ischemic episode causes severe changes in the corpus callosum and subcortical white matter [32,43,44,46,47,48]. These alterations are consistent with the activation of astrocytes and microglia in the brain tissue after ischemia [13,19,49]. Late brain white matter atrophy in experimental animals manifested as progressive spongiosis. Cerebral ischemic lesions showed signs of progressive neurodegeneration that developed slowly over a long period of time in post-ischemic survival [32]. A brain autopsy 1–2 years after experimental ischemia revealed cerebral hydrocephalus with the widening of the ventricles and the subarachnoid space [31,32,46,47]. This was accompanied by the complete atrophy of the hippocampus with a very narrow cortex [31,32,46,47,50]. The ultimate consequence of this neuropathology was the development of dementia in experimental and clinical studies after transient and reversible cerebral ischemia [45,47,51,52,53,54,55,56].
It has been found that neurodegenerative processes develop not only in the acute stage of ischemia, but also progress throughout the survival period after ischemia [32]. Brain neurodegeneration seen after ischemia shares features and mechanisms with neurodegeneration seen in Alzheimer’s disease [5,8,14,15,25,47,48,57,58,59,60,61]. This is confirmed by the increased permeability of the blood–brain barrier after ischemia to inflammatory cells and the leakage of amyloid and tau protein from the blood serum into the brain parenchyma, which in turn probably leads to irreversible and progressive damage to the whole brain [13,19,43,44,55,56,57,62,63,64,65,66,67,68,69,70,71,72,73,74,75]. Mental health deterioration and the onset of post-ischemic neurodegeneration-related cognitive impairment have raised concerns and triggered serious scientific debate. Progressive ischemic neurodegeneration of the brain has been found to be associated with the overproduction of folding proteins such as amyloid and tau protein [5,6,7,47,48,57,59,60,76,77,78,79,80]. Diffuse and senile amyloid plaques were found in the hippocampus and brain cortex after ischemia [57,81,82,83,84,85,86]. Data indicate that cerebral ischemia is involved in the development of paired helical filaments [87], neurofibrillary tangle-like [88,89,90], and neurofibrillary tangles [91,92] after this event. In this regard, special attention has been paid to the role of amyloid and tau protein as additional new contributing factors in the development of post-ischemic dementia [6,7,8,25,55,56,59]. Recently, changes in proteins and genes associated with Alzheimer’s disease following ischemic brain injury and their possible role in ischemic brain neurodegeneration were presented. New advances in understanding the development of post-ischemic neurodegeneration have revealed a dysregulation of the genes for amyloid protein precursor, α-secretase, β-secretase, presenilin 1 and 2, tau protein, autophagy, mitophagy, and apoptosis [7,16,17,18,93,94,95,96]. Ultimately, Alzheimer’s disease-related proteins and their genes have been documented to play an important role in the progression of post-ischemic brain neurodegeneration with subsequent development of dementia [8].
Due to the loss of neurons in the brain and the associated loss of neuronal network function, brain ischemia is also the leading cause of permanent disability in adults worldwide, reducing patient quality of life and increasing the global medical burden [2]. Early neuropsychiatric symptoms and dementia following ischemic stroke increase the risk of mortality and recurrence of ischemic stroke [97,98,99]. Despite its enormous impact on the socio-economic development of countries, this growing problem has so far received very little attention. Current management and treatment of most of the sequelae of ischemic stroke are unsatisfactory, with the exception of some antidepressants, which have therapeutic benefit [1].
In the absence of a translation of experimental neuroprotective substances for clinical use [100], the patients are very interested in improving motor and cognitive functions after ischemia, and not in protecting ischemic neuronal death in the brain. Therefore, we are now forced to improve the survival of persistent neurons and the associated cognitive functions after ischemia [27,51,52,53,54]. New treatments should improve activity following ischemic brain injury via effectively extending the therapeutic window. It should be emphasized that, despite the fact that ischemic stroke is one of the leading causes of death and disability worldwide, there has recently been a lack of effective post-stroke causal therapies that heal the structural and functional injuries, ultimately leading to post-ischemic neurodegeneration of the brain with subsequent developmental dementia. Therefore, in this review we will focus on the protective effect of pleiotropic myricetin on persistent neurons and neuropathological phenomena that develop after ischemic stroke and experimental cerebral ischemia.
2. Myricetin
3,3′,4′,5,5′,7-hexahydroxyflavone (myricetin) (Figure 1) is a flavonoid that was first identified in Myricaceae plants about 2 centuries ago [101,102]. Myricetin is light yellow in color. Commonly consumed fruits and vegetables are rich in myricetin. Strawberries, spinach, apples, aloe, carrots and mulberries are rich in myricetin, and the myricetin content in red wine is twice that of resveratrol [103,104,105,106]. In addition, myricetin has been approved as a health product in US by Food and Drug Administration and in Europe and has been successfully introduced to the general market [107]. Moreover, myricetin is an essential component of healthy food and drink and has an excellent safety profile combined with the possibility of human consumption [101]. Being one of the more studied polyphenols, myricetin has a number of biological properties [102]. Myricetin has been shown to have anti-oxidant, anti-inflammatory and anti-tumor effects [102,108,109]. Myricetin has been found to have therapeutic properties in Alzheimer’s disease [102]. It is believed that aloe and mulberry rich in myricetin have additional anti-dementia effects [102,110,111].
3. Possible Use of Myricetin in Post-Ischemic Neurodegeneration
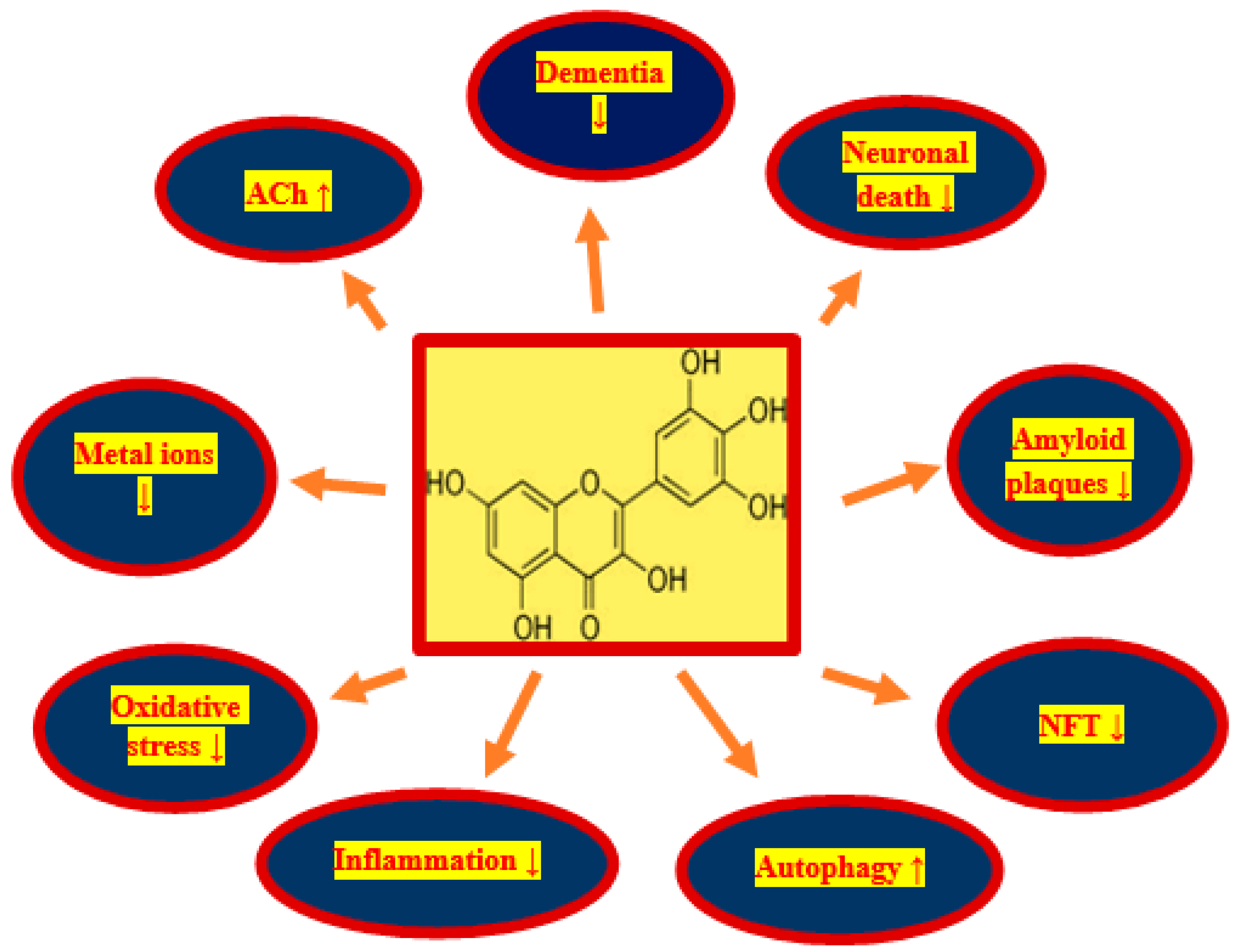
Transient ischemic-reperfusion brain injury in humans and animals is predominantly age-related and is characterized by various cognitive impairments, but primarily memory deficits, with gradual cognitive and intellectual decline, ultimately leading to the full-blown dementia [27,51,52,53,54]. In elderly humans, it is the leading cause of death. The accumulation of diffuse and senile amyloid plaques in the brain and neurofibrillary tangles in neuronal cells are new phenomenona in patients with cerebral ischemia and in animals post-ischemia [57,81,82,83,84,85,86,87,88,89,90,91,92]. It is believed that the deposition of misfolded proteins is an additional cause of neuronal death, loss of synapses, oxidative damage, and the development of neuroinflammation in the brain following ischemia [13,19,35,36,47,48]. Therefore, the greatest medicinal potential for neurodegeneration after stroke and animal ischemia should have molecules with pleiotropic activity, especially those with anti-amyloid, anti-tau protein and anti-dementia properties, as well as those that reduce oxidative stress and neuroinflammation [102]. Recently, myricetin is considered to be one of the most interesting and promising natural pleiotropic substance for use in the treatment of stroke due to its pleiotropic effects [102]. Myricetin is a substance with strong anti-oxidant and anti-inflammatory properties [102]. Its anti-amyloid and anti-tau protein properties make it the most promising compound for the treatment of various neurodegenerative disorders involving the deposition of folding proteins [102]. Neuroinflammation, oxidative damage, and misfolded protein deposition have been shown to synergistically contribute to post-ischemic brain injury [13,19,57,81,82,83,84,85,86,87,88,89,90,91,92]. Therefore, targeting these phenomena may be an essential strategy in the treatment of post-ischemic brain neurodegeneration. In this context, the use of myricetin in the treatment of ischemic neurodegeneration has certain advantages: namely, it (1) decreases the production of amyloid, (2) prevents the development of amyloid oligomers and fibrils, (3) protects the development of neurofibrillary tangles, (4) decreases neuroinflammation, (5) acts as a powerful anti-oxidant, (6) prevents metals from binding to amyloid and tau protein, (7) increases acetylcholine levels (Figure 1), and (8) can be taken in relatively high quantities without side effects [102].
Brain injury following ischemia is a neurodegenerative disease that can cause patients to gradually lose their ability to live independently and change their personality and behavior. Survivors of cerebral ischemia suffer from cognitive and linguistic deterioration, including speech and memory. Ischemic stroke not only threatens the life and health of patients, but also causes serious social problems, especially in countries with an aging population [2]. Recently, neuropsychiatric problems and post-stroke dementia have been of concern to clinicians and researchers. Mental problems and dementia are common post-stroke complications and are associated with poorer outcomes, including poor quality of life, increased burden of care, and poor functional status [112,113]. Studies have shown that myricetin ameliorates cognitive dysfunction by promoting amyloid clearance and inhibiting neuroinflammation in animal models of Alzheimer’s disease [110,111,114]. In this section of the review, we present a possible use of myricetin in the treatment of post-ischemic brain neurodegeneration that has neuropathological changes similar to Alzheimer’s disease.
4. Myricetin versus Amyloid
Following cerebral ischemia, β- and γ-secretase are involved in the production of amyloid from the amyloid protein precursor [93,94,95,96]. The overproduction and increase deposition of amyloid in the brain following ischemia [31,32,57] are associated with the onset and progression of neuropathological changes characteristic of Alzheimer’s disease. It has been shown that myricetin inhibits the activity of β-secretase, thus decreasing the production of amyloid [115]. Moreover, myricetin has been shown to increase the level of α-secretase, which results in an increased level of harmless fragments of the amyloid protein precursor [115]. This results in an overall decrease in the levels of the amyloid protein precursor that is used for amyloid production, thereby indirectly decreasing amyloid generation, too. The amyloid monomer exhibits neurotrophic activity, but amyloid oligomers and fibrils show strong neurotoxicity through increased neuroinflammation, destruction of cell membranes and oxidative stress [116,117,118]. Amyloid oligomers and fibrils are produced by excess amyloid through the β-sheet [116], and myricetin inhibits β-sheet formation [102,115,119]. Moreover, myricetin may bind to amyloid fibrils, thus inhibiting amyloid fibril elongation and maturation and the formation of diffuse and senile amyloid plaques (Figure 1) [102,120,121]. Thus, myricetin hinders the formation of amyloid oligomers, which decreases the neurotoxicity of amyloid and decreases the progression of ischemic pathological damage.
5. Myricetin versus Autophagy
Autophagy takes place within lysosomes to eliminate worn-out organelles or damaged proteins [41]. Autophagy and mitophagy proteins and gene alterations are observed in post-ischemic brain neurodegeneration [8,16,17,18]. In the case of neurons, it is difficult to get rid of, for example, toxic proteins (amyloid, dysfunctional tau protein) by cell division, so autophagy is a particularly important process for these cells. At the beginning of the autophagy, the endoplasmic reticulum in the neuron forms autophagic vesicles. These autophagic vesicles surround harmful proteins such as amyloid and incorrectly phosphorylated tau protein to form autophagosomes. Then the autophagosomes are transported along the microtubules by kinesin to the lysosomes, where the degradation of toxic proteins by lysosomes takes place [42,122]. In addition, current studies indicate that mammalian targets of rapamycin are a key signaling factors controlling cell proliferation and apoptosis and a major cytokine increasing cellular autophagy. Myricetin influences the increase of autophagy (Figure 1), which results in the elimination of toxic amyloid and the dysfunctional tau protein produced by neurons [102,123]. Myricetin induces protective autophagy by inhibiting the phosphorylation of mammalian targets of rapamycin, and this effect is dose dependent [102,123].
6. Myricetin versus Metal Ions
Disruption of the balance of metal ions in an ischemic brain can result in cytotoxicity, oxidative stress, and increased amyloid deposition—changes closely related to post-ischemic brain neurodegeneration [9,10,11,14]. Initially, studies on metals associated with ischemia focused on calcium ions [9], but in recent years attention has been focused on other metal ions such as zinc (Zn), copper (Cu), and iron (Fe), which are associated with the development of post-ischemic neurodegeneration [9]. Observations have shown that there are several binding sites within amyloid that bind metal ions, and a significant increase in amyloid toxicity is seen when this complexation occurs. Consider Zn2+ which has binding sites in amyloid, so even at micromolar concentrations it increases amyloid aggregation [124]. Myricetin has the ability to chelate metal ions, which may inhibit the effects of ischemia by controlling the concentration of metal ions in the brain [125]. It has been shown that myricetin can inhibit amyloid aggregation through chelation of Cu2+ or Zn2+ [126]. Myricetin can control the level of metal ions in an ischemic brain by forming complexes with metal ions, which decreases the likelihood of amyloid binding to metal ions. Moreover, myricetin may not only prevent binding of amyloid to metal ions, but also break down the resulting Zn2+/Cu2+ amyloid complexes [126]. In addition to affecting amyloid aggregation, Zn2+ may also have an effect on amyloid generation (Figure 1). Zinc can increase the level of β- and γ-secretase by inhibiting the activity of α-secretase, thus affecting the level of generated amyloid in the brain [127]. This indicates that the complexation of myricetin and Zn2+ may also decrease the level of amyloid. Moreover, like other divalent ions, Fe2+ can bind amyloid and trigger the development of amyloid oligomers and fibrils [128]. Upon complexation of Fe2+ by myricetin, the level of free Fe2+ decreases, which decreases the Fenton reaction converting H2O2 into highly toxic hydroxyl radicals and reactive oxygen species (ROS) [129,130]. This can lead to less damage from oxidative stress in an ischemic brain (Figure 1). Additionally, Fe2+ may lead to microglia activation, resulting in brain damage due to the development of neuroinflammation [131]. The effect of myricetin on Fe2+ may decreases the risk of neuroinflammation (Figure 1). Myricetin may also decrease iron levels by inhibiting the expression of the transferrin receptor 1 [132].
7. Myricetin versus Oxidative Stress
Oxidative stress is one of the direct mechanisms involved in the development of ischemic neurodegeneration of the brain [14]. Physiologically, the brain has high oxygen consumption but low antioxidant capacity, which makes it particularly susceptible to oxidative stress [133]. Free radicals and ROS are the two main mechanisms used by oxidative stress to injure neurons. By combining with radicals myricetin creates stable semiquinone radicals, breaking the radical chain reaction. In vitro experiments have shown that dose-dependent myricetin can effectively inhibit the formation of ROS and protect neurons from injury caused by oxidative stress (Figure 1) [134]. The effect of this is direct and indirect restoration of the physiological levels and activity of antioxidants such as catalase activity, superoxide dismutase (SOD), and glutathione peroxidase in cells [135,136]. Furthermore, in the H2O2-induced cell damage model, myricetin prevents oxidative stress-induced damage to DNA and lipids by regulating mitogen-activated protein kinase and phosphatidylinositol 3-kinase/protein kinase B signaling pathways. This leads to an increase in the level of anti-apoptotic molecules such as Bcl-2 and a decrease of pro-apoptotic mediators such as Bax, caspase-9, and caspase-3, resulting in inhibition of apoptosis induced by oxidative stress in cells (Figure 1) [135,137]. In addition to its direct effect on brain injury, oxidative stress is interrelated with factors associated with ischemic brain alteration, such as amyloid, which is capable of inducing oxidative stress, and oxidative stress induces amyloid production [138,139]. Myricetin inhibits the free radical chain reaction by inhibiting the amyloid source, thus decreasing the brain injury caused by oxidative stress [115]. By activating the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) pathway, oxidative stress increases the level of β-secretase. β-secretase metabolizes amyloid protein precursor to amyloid, leading to an increase in amyloid levels in ischemic brain [57,93,96], and an increase in amyloid levels further activates the JNK/SAPK pathway leading to a vicious cycle. Thus, myricetin may protect ischemic neurons from death due to its own antioxidant properties.
8. Myricetin versus Neuroinflammation
Investigations indicate that neuroinflammation is also one of the major processes of ischemic neurodegeneration, although neuroinflammation is to be the result of ischemic brain damage [13,19]. Myricetin directly decreases neuroinflammatory intensification (Figure 1), by inhibiting microglia activation and nucleotide-binding oligomerization domain-like receptor protein 3 [102,140]. Ultimately, these anti-inflammatory effects of myricetin help to ameliorate the severity of post-ischemic pathology in the brain (Figure 1). Myricetin can decrease the level of inflammatory factors such as interleukin (IL), tumor necrosis factor-alpha (TNF-α), and nuclear factor kappa B (NF-κB) [136,140]. Considering interleukin-1 (IL-1), it can not only damage neurons, but also elevate the level of the amyloid protein precursor, which increases the generation and accumulation of amyloid in the brain [141]. On top of it, IL-1 also increases TNF-α, which exacerbates the neuroinflammatory response and increase the cell and brain damage [141]. The anti-inflammatory activity of myricetin is mainly by decreasing the levels of IL, TNF-α, inducible nitric oxide synthase and cyclooxygenase-2 in the brain by disrupting the NF-κB signaling pathway and mitigating the damage caused by these factors [140].
Activated microglia play an important role in the development of post-ischemic neuroinflammation in the brain tissue [13,19]. Activated microglia occurs in two forms, M1 and M2: M1 promotes the development of neuroinflammation of the brain, while M2 inhibits the development of neroinflammation [142]. Finally, in the pathological process, the nucleotide-binding oligomerization domain-like receptor protein 3 in the brain can be activated by amyloid which can exacerbate neuropathological processes [143]. Studies show that amyloid levels can be effectively lowered by inhibiting nucleotide-binding oligomerization domain-like receptor protein 3, and that spatial memory impairment can be ameliorated in this situation (Figure 1) [143,144]. Currently, nucleotide-binding oligomerization domain-like receptor protein 3 in the brain is considered an effective target in the therapy of neurodegenerative disorders [145]. Moreover, myricetin may inhibit the activation of nucleotide-binding oligomerization domain-like receptor protein 3 by inhibiting apoptosis-associated speck-like proteins oxygen-dependent ubiquitination and promoting oxygen-independent ubiquitination of nucleotide-binding oligomerization domain-like receptor protein 3 [146]. The inhibitory effect of myricetin on nucleotide-binding oligomerization domain-like receptor protein 3 may decrease post-ischemic neuroinflammation and, to some extent, decrease amyloid levels in the brain (Figure 1).
9. Myricetin versus Acetylcholine
Acetylcholine is a neurotransmitter that plays a key role in the transmission of neural signals and memory formation, and the absence of acetylcholine in the brain, especially in the hippocampus is a known result of dementia in post-ischemic neurodegeneration [12]. Myricetin has been shown to be effective in inhibiting acetylcholinesterase, which breaks down acetylcholine in the brain [132,147]. Myricetin was effective in reducing the learning and memory impairment in aging brain through its ability to inhibit acetylcholinesterase (Figure 1) [148]. In addition to acetylcholinesterase, some inflammatory factors such as IL-1 also affect acetylcholine levels. IL-1 may increase the level of acetylcholinesterase and accelerate the breakdown of acetylcholine, causing insufficient content of acetylcholine in the brain and affecting the ability to remember [141]. The anti-inflammatory ability of myricetin may also indirectly prevent acetylcholine loss.
10. Myricetin versus Tau Protein
Disruption of the balance of metal ions in an ischemic brain can result in cytotoxicity, oxidative stress, increased amyloid deposition and tau protein hyperphosphorylation, changes closely related to post-ischemic brain neurodegeneration [9,10,11,14]. Zinc which has binding sites in tau protein, even at micromolar concentrations increases abnormal tau protein conformation (Figure 1) [124]. It has been shown that myricetin can inhibit tau protein hyperphosphorylation, through chelation of Zn2+ [126]. Oxidative stress also promotes tau protein hyperphosphorylation by inhibiting protein phosphatase 2A, and the dysfunctional tau protein increases oxidative stress and finally destroying synapses and mitochondria in a vicious circle [138,149,150,151]. IL-1 can also trigger tau protein hyperphosphorylation in the brain with further formation of neurofibrillary tangles, and neurofibrillary tangles are one of the major pathological elements of post-ischemic brain neurodegeneration in addition to amyloid (Figure 1) [91,92,141]. The anti-oxidative and anti-inflammatory ability of myricetin may also indirectly prevent tau protein hyperphosphorylation.
11. Myricetin versus Experimental Brain Ischemia
The aim of preliminary preclinical studies was to investigate whether myricetin may prevent damage from local brain ischemia and to identify potential mechanisms involved [136,152]. The rate of neurological deficit and the area of infarction caused by focal brain ischemia decreased in a dose-dependent manner after treatment with myricetin [136,152]. In addition, myricetin decreased the levels of interleukin-6, interleukin-1β, malondialdehyde, and TNF-α and increased the glutathione/glutathione disulfide ratio and SOD action [136]. In response to myricetin, a significant decrease in neuronal death was observed [136,152]. Moreover, myricetin significantly increased the level of phosphorylated protein kinase B (AKT) and decreased the phosphorylation of p38 mitogen-activated protein kinase (p38 MAPK) and the level of nuclear factor kappa B/p65 (NF-κB/p65) [136]. Taken together, the observations from these studies suggest that myricetin has a neuroprotective effect by decreasing ischemic brain damage via enhancement of the activity of antioxidant enzymes and improvement of mitochondrial function and that the protective effect of myricetin may be related to inactivation of the p38 MAPK and NF-κB/p65 pathways and activation of the AKT and nuclear factor erythroid 2-related factor 2 pathways [136,152].
12. Conclusions
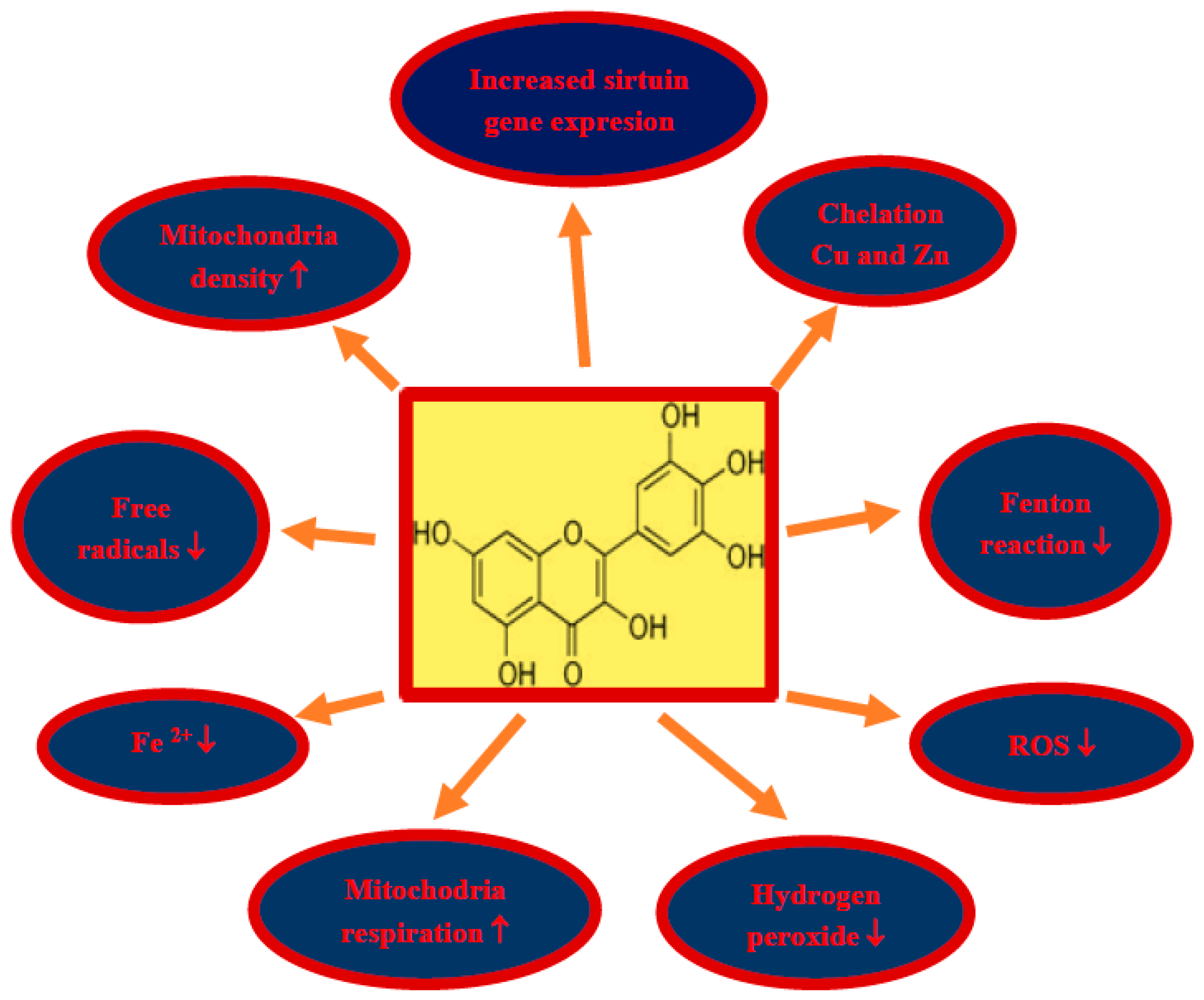
Damage and loss of neurons, with the accumulation of misfolded proteins in the form of amyloid plaques and neurofibrillary tangles, as well as impaired motor activity with the development of full-blown Alzheimer’s disease dementia are the main phenomena in brain neurodegeneration after ischemia in humans and animals. Due to the pleiotropic effects of myricetin, including anti-amyloid, anti-tau protein, anti-oxidant, anti-inflammatory and anti-dementia properties, myricetin is a promising candidate for the treatment of neurodegeneration following cerebral ischemia (Figure 1). Above effects partially can be explained by increases in sirtuin 1, sirtuin 3, and sirtuin 5 expression in mice treated by myricetin (Figure 2) [153]. Additionally, in this study it was found that myricetin increased mitochondrial content, respiration, and metabolism, and regulated ATP synthesis and cellular ATP generation by an increase in sirtuin 3 activity [153]. The main myricetin molecular mechanisms involved in the protection of neurons by myricetin in post-ischemic brain neurodegeneration are summarized in Figure 2. In addition, it is a safe substance, approved as a pro-health substance, commercially available and inexpensive, which can effectively cross the blood–brain barrier [154]. Holland et al. [154] decided to support the above view to determine whether dietary intake of myricetin in different doses (0.14–1.37 mg/d) by humans is associated with Alzheimer’s disease development and dementia. They presented that the onset of Alzheimer’s disease is inversely associated with dietary intake of myricetin [154]. Higher dietary intake of myricetin may be associated with decreased risk by 38% of the development of Alzheimer’s disease dementia in patients [154]. Recapitulating, the information available in this article about the therapeutic potential of myricetin provides significant evidence for the potential clinical utility of myricetin in the treatment of neurodegenerative disorders with misfolding proteins including post-ischemic brain neurodegeneration [154].
Author Contributions
Conceptualization, R.P.; methodology, S.J.; validation, R.P. and S.J.C.; writing—original draft preparation, R.P.; writing—review and editing, S.J.C; visualization, S.J.; supervision, R.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the financial support from the following institutions: the Mossakowski Medical Research Institute, Polish Academy of Sciences, Warsaw, Poland (T3-RP) and the Medical University of Lublin, Lublin, Poland (DS 475/20-SJC).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, S.; Xu, M.; Liu, Z.J.; Feng, J.; Ma, Y. Neuropsychiatric issues after stroke: Clinical significance and therapeutic implications. World J. Psychiatr. 2020, 10, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634. [Google Scholar] [CrossRef] [PubMed]
- Owolabi, M.O.; Akarolo-Anthony, S.; Akinyemi, R.; Arnett, D.; Gebregziabher, M.; Jenkins, C.; Tiwari, H.; Arulogun, O.; Akpalu, A.; Sarfo, F.S.; et al. Members of the H3 Africa Consortium. Members of the H3 Africa Consortium. The burden of stroke in Africa: A glance at the present and a glimpse into the future. Cardiovasc. J. Afr. 2015, 26 (Suppl. 1), S27–S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejot, Y.; Daubail, B.; Giroud, M. Epidemiology of stroke and transient ischemic attacks: Current knowledge and perspectives. Rev. Neurol. 2016, 172, 59–68. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S. Amyloid pathology in the brain after ischemia. Folia Neuropathol. 2019, 57, 220–226. [Google Scholar] [CrossRef]
- Pluta, R.; Bogucka-Kocka, A.; Ułamek-Kozioł, M.; Bogucki, J.; Czuczwar, S.J. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2018, 70, 881–884. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Tau protein dysfunction after brain ischemia. J. Alzheimer’s Dis. 2018, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R. Brain Ischemia: Alzheimer’s Disease Mechanisms; Nova, Science Publishers, Inc.: New York, NY, USA, 2019; p. 311. [Google Scholar]
- Pluta, R.; Salinska, E.; Puka, M.; Stafiej, A.; Łazarewicz, J.W. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, B.; Yin, L.; Cai, B.; Shan, H.; Zhang, L.; Lu, Y.; Bi, Z. Tanshinone IIA prevented brain iron dyshomeostasis in cerebral ischemic rats. Cell Physiol. Biochem. 2011, 27, 23–30. [Google Scholar] [CrossRef]
- Pochwat, B.; Nowak, G.; Szewczyk, B. Relationship between Zinc (Zn (2+) ) and glutamate receptors in the processes underlying neurodegeneration. Neural. Plast. 2015, 2015, 591563. [Google Scholar] [CrossRef]
- Yuan, Y.; Shan, X.; Men, W.; Zhai, H.; Qiao, X.; Geng, L.; Li, C. The effect of crocin on memory, hippocampal acetylcholine level, and apoptosis in a rat model of cerebral ischemia. Biomed. Pharmacother. 2020, 130, 110543. [Google Scholar] [CrossRef] [PubMed]
- Sekeljic, V.; Bataveljic, D.; Stamenkovic, S.; Ułamek, M.; Jabłoński, M.; Radenovic, L.; Pluta, R.; Andjus, P.R. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 2012, 217, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Furmaga-Jabłonska, W.; Maciejewski, R.; Ułamek-Kozioł, M.; Jabłoński, M. Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in sporadic Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Jabłonski, M.; Ułamek-Kozioł, M.; Kocki, J.; Brzozowska, J.; Januszewski, S.; Furmaga-Jabłonska, W.; Bogucka-Kocka, A.; Maciejewski, R.; Czuczwar, S.J. Sporadic, Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol. Neurobiol. 2013, 48, 500–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Furmaga-Jabłońska, W.; Brzozowska, J.; et al. Dysregulation of autophagy, mitophagy and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 54, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of autophagy, mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J. Alzheimer’s Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging 2020, 12, 12251–12267. [Google Scholar] [CrossRef]
- Battaglini, D.; Pimentel-Coelho, P.M.; Robba, C.; Dos Santos, C.C.; Cruz, F.F.; Pelosi, P.; Rocco, P.R.M. Gut microbiota in acute ischemic stroke: From pathophysiology to therapeutic implications. Front. Neurol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Tan, B.Y.Q.; Paliwal, P.R.; Sharma, V.K. Gut microbiota and stroke. Ann. Indian Acad. Neurol. 2020, 23, 155–158. [Google Scholar] [CrossRef]
- Sexton, E.; McLoughlin, A.; Williams, D.J.; Merriman, N.A.; Donnelly, N.; Rohde, D.; Hickey, A.; Wren, M.A.; Bennett, K. Systematic review and meta-analysis of the prevalence of cognitive impairment no dementia in the first year post-stroke. Eur. Stroke J. 2019, 4, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellon, L.; Brewer, L.; Hall, P.; Horgan, F.; Williams, D.; Hickey, A. Cognitive impairment six months after ischaemic stroke: A profile from the ASPIRE-S study. BMC Neurol. 2015, 15, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J.W.; Crawford, J.D.; Desmond, D.W.; Godefroy, O.; Jokinen, H.; Mahinrad, S.; Bae, H.J.; Lim, J.S.; Kohler, S.; Douven, E.; et al. Stroke and Cognition (STROKOG) Collaboration. Profile of and risk factors for post-stroke cognitive impairment in diverse ethno-regional groups. Neurology 2019, 93, e2257–e2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R. Ischemia-Reperfusion Pathways in Alzheimer’s Disease; Nova, Science Publishers, Inc.: New York, NY, USA, 2007. [Google Scholar]
- Pluta, R.; Jolkkonen, J.; Cuzzocrea, S.; Pedata, F.; Cechetto, D.; Popa-Wagner, A. Cognitive impairment with vascular impairment and degeneration. Curr. Neurovasc. Res. 2011, 8, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, Y. Dementia and death after stroke in older adults during a 10-year follow-up: Results from a competing risk model. J. Nutr. Health Aging 2018, 22, 297–301. [Google Scholar] [CrossRef]
- Kuzma, E.; Lourida, I.; Moore, S.F.; Levine, D.A.; Ukoumunne, O.C.; Llewellyn, D.J. Stroke and dementia risk: A systematic review and meta-analysis. Alzheimer’s Dement. 2018, 14, 1416–1426. [Google Scholar] [CrossRef] [Green Version]
- De Ronchi, D.; Palmer, K.; Pioggiosi, P.; Atti, A.R.; Berardi, D.; Ferrari, B.; Dalmonte, E.; Fratiglioni, L. The combined effect of age, education, and stroke on dementia and cognitive impairment no dementia in the elderly. Dement. Geriatr. Cogn. Disord. 2007, 24, 266–273. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and metaanalysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Pluta, R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia–reperfusion brain injury. A model of early Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 903, 324–334. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek, M.; Jabłoński, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. 2009, 292, 1863–1881. [Google Scholar] [CrossRef]
- Gemmell, E.; Bosomworth, H.; Allan, L.; Hall, R.; Khundakar, A.; Oakley, A.E.; Deramecourt, V.; Polvikoski, T.M.; O’Brien, J.T.; Kalaria, R.N. Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke 2012, 43, 808–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemmell, E.; Tam, E.; Allan, L.; Hall, R.; Khundakar, A.; Oakley, A.E.; Thomas, A.; Deramecourt, V.; Kalaria, R.N. Neuron volumes in hippocampal subfields in delayed poststroke and aging-related dementias. J. Neuropathol. Exp. Neurol. 2014, 73, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Shared genomic and proteomic contribution of amyloid and tau protein characteristic of Alzheimer’s disease to brain ischemia. Int. J. Mol. Sci. 2020, 21, 3186. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Participation of amyloid and tau protein in neuronal death and neurodegeneration after brain ischemia. Int. J. Mol. Sci. 2020, 21, 4599. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.W.; Matsumoto, K.; Li, H.B.; Murakami, Y.; Watanabe, H. Neuronal damage and decrease of central acetylcholine level following permanent occlusion of bilateral common carotid arteries in rat. Brain Res. 1995, 673, 290–296. [Google Scholar] [CrossRef]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2013, 14, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.W.; Han, X.J.; Shi, Z.S.; Lei, Z.G.; Xu, Z.C. Remodeling of synapses in the CA1 area of the hippocampus after transient global ischemia. Neuroscience 2012, 218, 268–277. [Google Scholar] [CrossRef]
- Hofmeijer, J.; van Putten, M.J. Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Furmaga-Jabłońska, W.; Januszewski, S.; Brzozowska, J.; Sciślewska, M.; Jabłoński, M.; Pluta, R. Neuronal autophagy: Self-eating or self-cannibalism in Alzheimer’s disease. Neurochem. Res. 2013, 38, 1769–1773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cao, Y.; Liu, C. Autophagy and ischemic stroke. Adv. Exp. Med. Biol. 2020, 1207, 111–134. [Google Scholar]
- Pluta, R.; Ułamek, M.; Januszewski, S. Micro-blood–brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Acta Neurochir. 2006, 96, 267–271. [Google Scholar]
- Pluta, R.; Januszewski, S.; Ułamek, M. Ischemic blood–brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. Acta Neurochir. 2008, 102, 353–356. [Google Scholar]
- Scherr, M.; Trinka, E.; McCoy, M.; Krenn, Y.; Staffen, W.; Kirschner, M.; Bergmann, H.J.; Mutzenbach, J.S. Cerebral hypoperfusion during carotid artery stenosis can lead to cognitive deficits that may be independent of white matter lesion load. Curr. Neurovasc. Res. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Jabłonski, M.; Maciejewski, R.; Januszewski, S.; Ułamek, M.; Pluta, R. One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol. Res. 2011, 60 (Suppl. 1), 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Substantiation for the use of curcumin during the development of neurodegeneration after brain ischemia. Int. J. Mol. Sci. 2020, 21, 517. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Proteomic and genomic changes in tau protein, which are associated with Alzheimer’s disease after ischemia-reperfusion brain injury. Int. J. Mol. Sci. 2020, 21, 892. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, K.; Adachi, K.; Kataoka, S.; Watanabe, A.; Tabira, T.; Takahashi, K.; Wakita, H. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp. Neurol. 2008, 210, 585–591. [Google Scholar] [CrossRef]
- Hossmann, K.A.; Schmidt-Kastner, R.; Ophoff, B.G. Recovery of integrative central nervous function after one hour global cerebro-circulatory arrest in normothermic cat. J. Neurol. Sci. 1987, 77, 305–320. [Google Scholar] [CrossRef]
- De la Tremblaye, P.B.; Plamondon, H. Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav. Brain Res. 2011, 219, 227–233. [Google Scholar] [CrossRef]
- Kiryk, A.; Pluta, R.; Figiel, I.; Mikosz, M.; Ułamek, M.; Niewiadomska, G.; Jabłoński, M.; Kaczmarek, L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav. Brain Res. 2011, 219, 1–7. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.J.; Zhang, M.; Fang, C.Q.; Zhou, H.D. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci. 2011, 89, 86–92. [Google Scholar] [PubMed]
- Cohan, C.H.; Neumann, J.T.; Dave, K.R.; Alekseyenko, A.; Binkert, M.; Stransky, K.; Lin, H.W.; Barnes, C.A.; Wright, C.B.; Perez-Pinzon, M.A. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS ONE 2015, 10, e0124918. [Google Scholar]
- Liu, W.; Wong, A.; Au, L.; Yang, J.; Wang, Z.; Leung, E.Y.; Chen, S.; Ho, C.L.; Mok, V.C. Influence of amyloid-beta on cognitive decline after stroke/transient ischemic attack: Three-year longitudinal study. Stroke 2015, 46, 3074–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.H.; Cao, H.Y.; Wang, Y.R.; Jiao, S.S.; Bu, X.L.; Zeng, F.; Wang, Q.H.; Li, J.; Deng, J.; Zhou, H.D.; et al. Aβ is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox. Res. 2015, 27, 292–299. [Google Scholar] [PubMed]
- Pluta, R.; Kida, E.; Lossinsky, A.S.; Golabek, A.A.; Mossakowski, M.J.; Wisniewski, H.M. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994, 649, 323–328. [Google Scholar] [PubMed]
- Kida, E.; Pluta, R.; Lossinsky, A.S.; Golabek, A.A.; Choi-Miura, N.H.; Wisniewski, H.M.; Mossakowski, M.J. Complete cerebral ischemia with short-term survival in rat induced by cardiac arrest: II. Extracellular and intracellular accumulation of apolipoproteins E and J in the brain. Brain Res. 1995, 674, 341–346. [Google Scholar]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Common proteomic and genomic contribution to ischemic brain damage and Alzheimer’s disease. In Alzheimer’s Disease; Wisniewski, T., Ed.; Codon Publications: Brisbane, Australia, 2019; pp. 53–68. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M. The role of degenerative pathways in the development of irreversible consequences after brain ischemia. Neural. Regen. Res. 2019, 14, 982–983. [Google Scholar] [CrossRef]
- Pluta, R.; Lossinsky, A.S.; Wisniewski, H.M.; Mossakowski, M.J. Early blood–brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52. [Google Scholar]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wisniewski, H.M.; Mossakowski, M.J. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Hirnforsch. 1994, 35, 463–471. [Google Scholar]
- Wisniewski, H.M.; Pluta, R.; Lossinsky, A.S.; Mossakowski, M.J. Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol. 1995, 90, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Barcikowska, M.; Januszewski, S.; Misicka, A.; Lipkowski, A.W. Evidence of blood–brain barrier permeability/leakage for circulating human Alzheimer’s β-amyloid-(1–42)-peptide. Neuroreport 1996, 7, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Barcikowska, M.; Misicka, A.; Lipkowski, A.W.; Spisacka, S.; Januszewski, S. Ischemic rats as a model in the study of the neurobiological role of human β-amyloid peptide. Time-dependent disappearing diffuse amyloid plaques in brain. Neuroreport 1999, 10, 3615–3619. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Misicka, A.; Barcikowska, M.; Spisacka, S.; Lipkowski, A.W.; Januszewski, S. Possible reverse transport of β-amyloid peptide across the blood-brain barrier. Acta Neurochir. 2000, 76, 73–77. [Google Scholar]
- Pluta, R. Blood–brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia–reperfusion brain injury with 1-year survival. Acta Neurochir. 2003, 86, 117–122. [Google Scholar]
- Anfuso, C.D.; Assero, G.; Lupo, G.; Nicota, A.; Cannavo, G.; Strosznajder, R.P.; Rapisarda, P.; Pluta, R.; Alberghia, M. Amyloid beta(1-42) and its beta(25-35) fragment induce activation and membrane translocation of cytosolic phospholipase A(2) in bovine retina capillary pericytes. Biochim. Biophys. Acta 2004, 1686, 125–138. [Google Scholar] [CrossRef]
- Lee, P.H.; Bang, O.Y.; Hwang, E.M.; Lee, J.S.; Joo, U.S.; Mook-Jung, I.; Huh, K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural. Transm. 2005, 112, 1371–1379. [Google Scholar] [CrossRef]
- Pluta, R. Pathological opening of the blood–brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia–reperfusion brain injury. Chemotherapy 2005, 51, 223–226. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Jabłoński, M.; Ułamek, M. Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir. 2010, 106, 37–41. [Google Scholar]
- Mörtberg, E.; Zetterberg, H.; Nordmark, J.; Blennow, K.; Catry, C.; Decraemer, H.; Vanmechelen, E.; Rubertsson, S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 2011, 55, 1132–1138. [Google Scholar] [CrossRef]
- Zetterberg, H.; Mörtberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 2011, 6, e28263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, J.; Mörtberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2013, 84, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kida, E.; Golabek, A.A.; Mossakowski, M.J. Possible involvement of patomechanism(s) operating in brain ischemia, in Alzheimer’s disease pathology. J. Cereb. Blood Flow Metab. 1995, 15 (Suppl. 1), S803. [Google Scholar]
- Pluta, R. Proteins associated with Alzheimer’s disease in conditions predisposing to Alzheimer’s-type neurodegeneration. J. Cereb. Blood Flow Metab. 2001, 21 (Suppl. 1), S424. [Google Scholar]
- Badan, I.; Platt, D.; Kessler, C.; Popa-Wagner, A. Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology 2003, 49, 356–365. [Google Scholar] [CrossRef]
- Badan, I.; Dinca, I.; Buchhold, B.; Suofu, Y.; Walker, L.; Gratz, M.; Platt, D.; Kessler, C.H.; Popa-Wagner, A. Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur. J. Neurosci. 2004, 19, 2270–2280. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Dysregulation of Alzheimer’s disease-related genes and proteins following cardiac arrest. Folia Neuropathol. 2017, 55, 283–288. [Google Scholar] [CrossRef]
- Jendroska, K.; Poewe, W.; Daniel, S.E.; Pluess, J.; Iwerssen-Schmidt, H.; Paulsen, J.; Barthel, S.; Schelosky, L.; Cervos-Navarr, J.; DeArmond, S.J. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995, 90, 461–466. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Maslinska, D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996, 34, 65–71. [Google Scholar]
- Jendroska, K.; Hoffmann, O.M.; Patt, S. Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann. N. Y. Acad. Sci. 1997, 826, 401–405. [Google Scholar] [CrossRef]
- Van Groen, T.; Puurunen, K.; Maki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Wu, H.; Yang, Y.; Wand, D.; Chen, Y.; Gu, Y.; Liu, T. Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J. Alzheimer’s Dis. 2007, 12, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Akinyemi, R.O.; Allan, L.M.; Oakley, A.; Kalaria, R.N. Hippocampal neurodegenerative pathology in post-stroke dementia compared to other dementias and aging controls. Front. Neurosci. 2017, 11, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Yuldasheva, N.Y.; Batten, T.F.C.; Pickles, A.R.; Kellett, K.A.B.; Saha, S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia—A potential model for vascular dementia? Neurochem. Int. 2018, 118, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Yang, S.; Liu, R.; Simpkins, J.W. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004, 1022, 30–38. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Yang, S.H.; Liu, R.; Perez, E.J.; Brun-Ziukemagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta 2007, 1772, 473–483. [Google Scholar] [CrossRef]
- Kato, T.; Hirano, A.; Katagiri, T.; Sasaki, H.; Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 1988, 23, 620–623. [Google Scholar] [CrossRef]
- Hatsuta, H.; Takao, M.; Nogami, A.; Uchino, A.; Sumikura, H.; Takata, T.; Morimoto, S.; Kanemaru, K.; Adachi, T.; Arai, T.; et al. Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol. Commun. 2019, 7, 49. [Google Scholar] [CrossRef]
- Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Januszewski, S.; Jabłoński, M.; Gil-Kulik, P.; Brzozowska, J.; Petniak, A.; Furmaga-Jabłońska, W.; Bogucki, J.; et al. Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J. Alzheimer’s Dis. 2015, 47, 1047–1056. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Brzozowska, J.; Furmaga-Jabłońska, W.; et al. Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J Alzheimer’s Dis 2016, 51, 1023–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Gil-Kulik, P.; Januszewski, S.; Jabłoński, M.; Petniak, A.; Brzozowska, J.; Bogucki, J.; et al. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol. Rep. 2016, 68, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Kocki, J.; Bogucki, J.; Januszewski, S.; Bogucka-Kocka, A.; Czuczwar, S.J. Expression of the tau protein and amyloid protein precursor processing genes in the CA3 area of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. Mol. Neurobiol. 2020, 57, 1281–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, M.L.; Köhler, S.; O’Brien, J.T.; Mead, G.E. Neuropsychiatric outcomes of stroke. Lancet Neurol. 2014, 13, 525–534. [Google Scholar] [PubMed] [Green Version]
- Bartoli, F.; Di Brita, C.; Crocamo, C.; Clerici, M.; Carrà, G. Early post-stroke depression andmortality: Meta-analysis and meta-regression. Front. Psychiatry 2018, 9, 530. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Mueller, C.; Li, Y.J.; Shen, W.D.; Stewart, R. Post stroke depression and risk of stroke recurrence and mortality: A systematic review and meta-analysis. Aging Res. Rev. 2019, 50, 102–109. [Google Scholar] [CrossRef]
- Herson, P.S.; Traystman, R.J. Animal models of stroke: Translational potential at present and in 2050. Future Neurol. 2014, 9, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A dietary molecule with diverse biological activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Taheri, Y.; Suleria, H.A.R.; Martins, N.; Sytar, O.; Beyatli, A.; Yeskaliyeva, B.; Seitimova, G.; Salehi, B.; Semwal, P.; Painuli, S.; et al. Myricetin bioactive effects: Moving from preclinical evidence to potential clinical applications. BMC Complement. Med. Ther. 2020, 20, 241. [Google Scholar] [CrossRef]
- Lee, K.W.; Kang, N.J.; Rogozin, E.A.; Kim, H.G.; Cho, Y.Y.; Bode, A.M.; Lee, H.J.; Surh, Y.J.; Bowden, G.T.; Dong, Z. Myricetin is a novel natural inhibitor of neoplastic cell transformation and MEK1. Carcinogenesis 2007, 28, 1918–1927. [Google Scholar] [CrossRef]
- Rodrigo, R.; Miranda, A.; Vergara, L. Modulation of endogenous antioxidant system by wine polyphenols in human disease. Clin. Chim. Acta 2011, 412, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, T.; Anwar, F.; Abbas1, M.; Saari, N. Effect of maturity on phenolics (Phenolic acids and flavonoids) profile of strawberry cultivars and mulberry species from Pakistan. Int. J. Mol. Sci. 2012, 13, 4591–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.N.; Huang, H.Z.; Zhang, Q.L.; Fan, F.J.; Xu, C.J.; Sun, C.D.; Li, X.; Chen, K.S. Phytochemical characterization of chinese bayberry (Myrica Rubra Sieb. Et Zucc.) of 17 cultivars and their antioxidant properties. Int. J. Mol. Sci. 2015, 16, 12467–12481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, M.W. Anti-inflammatory activity of a complementary medicine, FYI™. Prog. Nutr. 2002, 4, 55–61. [Google Scholar]
- Zhang, D.; Xie, L.; Jia, G.; Cai, S.; Ji, B.; Liu, Y.; Wu, W.; Zhou, F.; Wang, A.; Chu, L.; et al. Comparative study on antioxidant capacity of flavonoids and their inhibitory effects on oleic acid-induced hepatic steatosis in vitro. Eur. J. Med. Chem. 2011, 46, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lindemeyer, A.K.; Gonzalez, C.; Shao, X.M.; Spigelman, I.; Olsen, R.W.; Liang, J. Dihydromyricetin as a novel anti-alcohol intoxication medication. J. Neurosci. 2012, 32, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clementinull, M.E.; Tringalinull, G.; Triggianinull, D.; Giardina, B. Aloe arborescens extract protects IMR-32 cells against Alzheimer amyloid beta peptide via inhibition of radical peroxide production. Nat. Prod. Commun. 2015, 10, 1993–1995. [Google Scholar]
- Liu, D.; Du, D. Mulberry fruit extract alleviates cognitive impairment by promoting the clearance of amyloid-β and inhibiting neuroinflammation in Alzheimer’s disease mice. Neurochem. Res. 2020, 45, 2009–2019. [Google Scholar] [CrossRef]
- Khan, A.; Chen, L.; Zhang, G.; Guo, X.; Wu, G.; Wang, H.; You, Y.; Gu, Y.; Yuan, Y. Management of poststroke neuropsychiatric disorders. Transl. Neurosci. Clin. 2016, 2, 244–251. [Google Scholar] [CrossRef]
- Stein, L.A.; Goldmann, E.; Zamzam, A.; Luciano, J.M.; Messé, S.R.; Cucchiara, B.L.; Kasner, S.E.; Mullen, M.T. Association between anxiety, depression, and post-traumatic stress disorder and outcomes after ischemic stroke. Front. Neurol. 2018, 9, 890. [Google Scholar] [CrossRef]
- Liang, J.; López-Valdés, H.E.; Martínez-Coria, H.; Lindemeyer, A.K.; Shen, Y.; Shao, X.M.; Olsen, R.W. Dihydromyricetin ameliorates behavioral deficits and reverses neuropathology of transgenic mouse models of Alzheimer’s disease. Neurochem. Res. 2014, 39, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Multifunction of myricetin on A beta: Neuroprotection via a conformational change of A beta and reduction of A beta via the interference of secretases. J. Neurosci. Res. 2008, 86, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Hyunbum, J.; Jie, Z.; Ruth, N. Models of beta-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007, 93, 1938–1949. [Google Scholar]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Lee, T.H.; Small, D.H.; Aguilar, M.I. Characterization of early stage intermediates in the nucleation phase of A beta aggregation. Biochemistry 2012, 51, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhao, W.; Sang, J.; Wang, W.; Wei, W.; Wang, Y.; Zhao, F.; Lu, F.; Liu, F. Inhibitory effect of a flavonoid dihydromyricetin against Aβ40 amyloidogenesis and its associated cytotoxicity. ACS Chem. Neurosci. 2019, 10, 4696–4703. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, M.; Hasegawa, K.; Tsutsumi-Yasuhara, S.; Ohhashi, Y.; Ookoshi, T.; Ono, K.; Yamada, M.; Naiki, H. The anti-amyloidogenic effect is exerted against Alzheimer’s β-amyloid fibrils in vitro by preferential and reversible binding of flavonoids to the amyloid fibril structure? Biochemistry 2007, 46, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Andarzi, G.S.; Barzegar, A.; Tarinejad, A. The role of phenolic OH groups of flavonoid compounds with H-bond formation ability to suppress amyloid mature fibrils by destabilizing β-sheet conformation of monomeric Aβ17-42. PLoS ONE 2018, 13, e0199541. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Cheng, Q.; Chen, C. The role of autophagy in the pathogenesis of ischemic stroke. Curr. Neuropharmacol. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Cao, J.; Chen, H.; Lu, W.; Wu, Y.; Wu, X.; Xia, D.; Zhu, J. Myricetin induces protective autophagy by inhibiting the phosphorylation of mTOR in HepG2 cells. Anat. Rec. (Hoboken) 2018, 301, 786–795. [Google Scholar] [CrossRef]
- Boom, A.; Authelet, M.; Dedecker, R.; Frédérick, C.; Van, H.R.; Daubie, V.; Leroy, K.; Pochet, R.; Brion, J.P. Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn2+ in human-tau transfected cells. Biochim. Biophys. Acta 2009, 1793, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Jomová, K.; Hudecova, L.; Lauro, P.; Simunkova, M.; Alwasel, S.H.; Alhazza, I.M.; Valko, M. A Switch between antioxidant and prooxidant properties of the phenolic compounds myricetin, morin, 3’,4’-dihydroxyflavone, taxifolin and 4-hydroxy-coumarin in the presence of copper(II) ions: A spectroscopic, absorption titration and DNA damage study. Molecules 2019, 24, 4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeToma, A.S.; Choi, J.S.; Braymer, J.J.; Lim, M.H. Myricetin: A naturally occurring regulator of metal-induced amyloid-β aggregation and neurotoxicity. ChemBioChem 2011, 12, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Garai, K.; Sengupta, P.; Sahoo, B.; Maiti, S. Selective destabilization of soluble amyloid β oligomers by divalent metal ions. Biochem. Biophys. Res. Commun. 2006, 345, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Kolandaivel, P. Fe(2+) binding on amyloid β-peptide promotes aggregation. Proteins 2016, 84, 1257–1274. [Google Scholar] [CrossRef]
- Armengou, A.; Davalos, A. A review of the state of research into the role of iron in stroke. J. Nutr. Health Aging 2002, 6, 207–208. [Google Scholar]
- Wang, T.; Xu, S.F.; Fan, Y.G.; Li, L.B.; Guo, C. Iron pathophysiology in Alzheimer’s diseases. Adv. Exp. Med. Biol. 2019, 1173, 67–104. [Google Scholar]
- Peters, D.G.; Pollack, A.N.; Cheng, K.C.; Sun, D.; Saido, T.; Haaf, M.P.; Yang, Q.X.; Connor, J.R.; Meadowcroft, M.D. Dietary lipophilic iron alters amyloidogenesis and microglial morphology in Alzheimer’s disease knock-in APP mice. Metallomics 2018, 10, 426–443. [Google Scholar] [CrossRef]
- Wang, B.; Zhong, Y.; Gao, C.; Li, J. Myricetin ameliorates scopolamine-induced memory impairment in mice via inhibiting acetylcholinesterase and down-regulating brain iron. Biochem. Biophys. Res. Commun. 2017, 490, 336–342. [Google Scholar] [CrossRef]
- Guo, H.; Cao, H.; Cui, X.; Zheng, W.; Wang, S.; Yu, J.; Chen, Z. Silymarin’s inhibition and treatment effects for Alzheimer’s disease. Molecules 2019, 24, 1748. [Google Scholar] [CrossRef] [Green Version]
- Barzegar, A. Antioxidant activity of polyphenolic myricetin in vitro cell-free and cell-based systems. Mol. Biol. Res. Commun. 2016, 5, 87–95. [Google Scholar] [PubMed]
- Wang, Z.H.; Ah, K.K.; Zhang, R.; Piao, M.J.; Jo, S.H.; Kim, J.S.; Kang, S.S.; Lee, J.S.; Park, D.H.; Hyun, J.W. Myricetin suppresses oxidative stress-induced cell damage via both direct and indirect antioxidant action. Environ. Toxicol. Pharmacol. 2010, 29, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xu, P.; Fu, T.; Huang, X.; Song, J.; Chen, M.; Tian, X.; Yin, H.; Han, J. Myricetin against ischemic cerebral injury in rat middle cerebral artery occlusion model. Mol. Med. Rep. 2018, 17, 3274–3280. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, J.; Lin, J.; Wang, T.; Huang, J.; Lin, Y.; Chen, D. Protective effects of dihydromyricetin against OH-induced mesenchymal stem cells damage and mechanistic chemistry. Molecules 2016, 21, 604. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 2, 4183–4187. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Galvan, V.; Lange, M.B.; Tang, H.; Sowell, R.A.; Spilman, P.; Fombonne, J.; Gorostiza, O.; Zhang, J.; Sultana, R.; et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice:Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic. Biol. Med. 2010, 48, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, N.; Li, X. Dihydromyricetin attenuates inflammation through TLR4/NF-kappaB pathway. Open Med. (Wars) 2019, 14, 719–725. [Google Scholar] [CrossRef]
- Shadfar, S.; Hwang, C.J.; Lim, M.S.; Choi, D.Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharm. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2012, 493, 674–678. [Google Scholar] [CrossRef]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 84, 875–882. [Google Scholar] [CrossRef]
- Saresella, M.; La, R.F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lin, H.; Xie, S.; Huang, B.; Qian, Y.; Chen, K.; Niu, Y.; Shen, H.M.; Cai, J.; Li, P.; et al. Myricetin inhibits NLRP3 inflammasome activation via reduction of ROS-dependent ubiquitination of ASC and promotion of ROS-independent NLRP3 ubiquitination. Toxicol. Appl. Pharmacol. 2019, 365, 19–29. [Google Scholar] [CrossRef]
- Pepeu, G.; Giovannini, M.G. Changes in acetylcholine extracellular levels during cognitive processes. Learn. Mem. 2004, 11, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, X.; Liu, X.; Chen, X.; Li, J.; Yang, X.; Fan, J.; Yang, Y.; Chen, N. Ampelopsin attenuates brain aging of D-gal-induced rats through miR-34a-mediated SIRT1/mTOR signal pathway. Oncotarget 2016, 7, 74484–74495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Chen, Q.B.; Xin, X.L.; Aisa, H.A. Anti-diabetic effect of three new norditerpenoid alkaloids in vitro and potential mechanism via PI3K/Akt signaling pathway. Biomed. Pharmacother. 2017, 87, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef]
- Akindehin, S.; Jung, Y.-S.; Kim, S.-N.; Son, Y.-H.; Lee, I.; Seong, J.K.; Jeong, H.W.; Lee, Y.-H. Myricetin exerts anti-obesity effects through upregulation of SIRT3 in adipose tissue. Nutrients 2018, 10, 1962. [Google Scholar] [CrossRef] [Green Version]
- Holland, T.M.; Agarwal, P.; Wang, Y.; Leurgans, S.E.; Bennett, D.A.; Booth, S.L.; Morris, M.C. Dietary flavonols and risk of Alzheimer dementia. Neurology 2020, 94, e1749–e1756. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Positive influence of myricetin on phenomena occurring in post-ischemic brain neurodegeneration. In a rectangle structure of myricetin, NFT-neurofibrillary tangles, ACh—acetylcholine, ↓—decrease, ↑—increase.
Figure 1.
Positive influence of myricetin on phenomena occurring in post-ischemic brain neurodegeneration. In a rectangle structure of myricetin, NFT-neurofibrillary tangles, ACh—acetylcholine, ↓—decrease, ↑—increase.

Figure 2.
Some molecular mechanisms involved in the protection of neurons by myricetin in post-ischemic brain neurodegeneration. In a rectangle structure of myricetin, ROS—reactive oxygen species, ↓—decrease, ↑—increase.
Figure 2.
Some molecular mechanisms involved in the protection of neurons by myricetin in post-ischemic brain neurodegeneration. In a rectangle structure of myricetin, ROS—reactive oxygen species, ↓—decrease, ↑—increase.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pluta, R.; Januszewski, S.; Czuczwar, S.J. Myricetin as a Promising Molecule for the Treatment of Post-Ischemic Brain Neurodegeneration. Nutrients 2021, 13, 342. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020342
AMA Style
Pluta R, Januszewski S, Czuczwar SJ. Myricetin as a Promising Molecule for the Treatment of Post-Ischemic Brain Neurodegeneration. Nutrients. 2021; 13(2):342. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020342
Chicago/Turabian StylePluta, Ryszard, Sławomir Januszewski, and Stanisław J. Czuczwar. 2021. "Myricetin as a Promising Molecule for the Treatment of Post-Ischemic Brain Neurodegeneration" Nutrients 13, no. 2: 342. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020342
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.