
Chk1 and the Host Cell DNA Damage Response as a Potential Antiviral Target in BK Polyomavirus Infection

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Immunohistochemical and Immunofluorescent Analysis of Biopsies
2.3. Immunofluorescent Staining of Cultured Cells
2.4. Laser Scanning Cytometry (LSC)
2.5. Immunoblotting
2.6. Alamar Blue Assay
2.7. siRNA-Mediated Knockdown
2.8. Quantification of Viral DNA
2.8.1. DNA Isolation from Cells and Electrophoresis
2.8.2. Real-Time PCR
3. Results
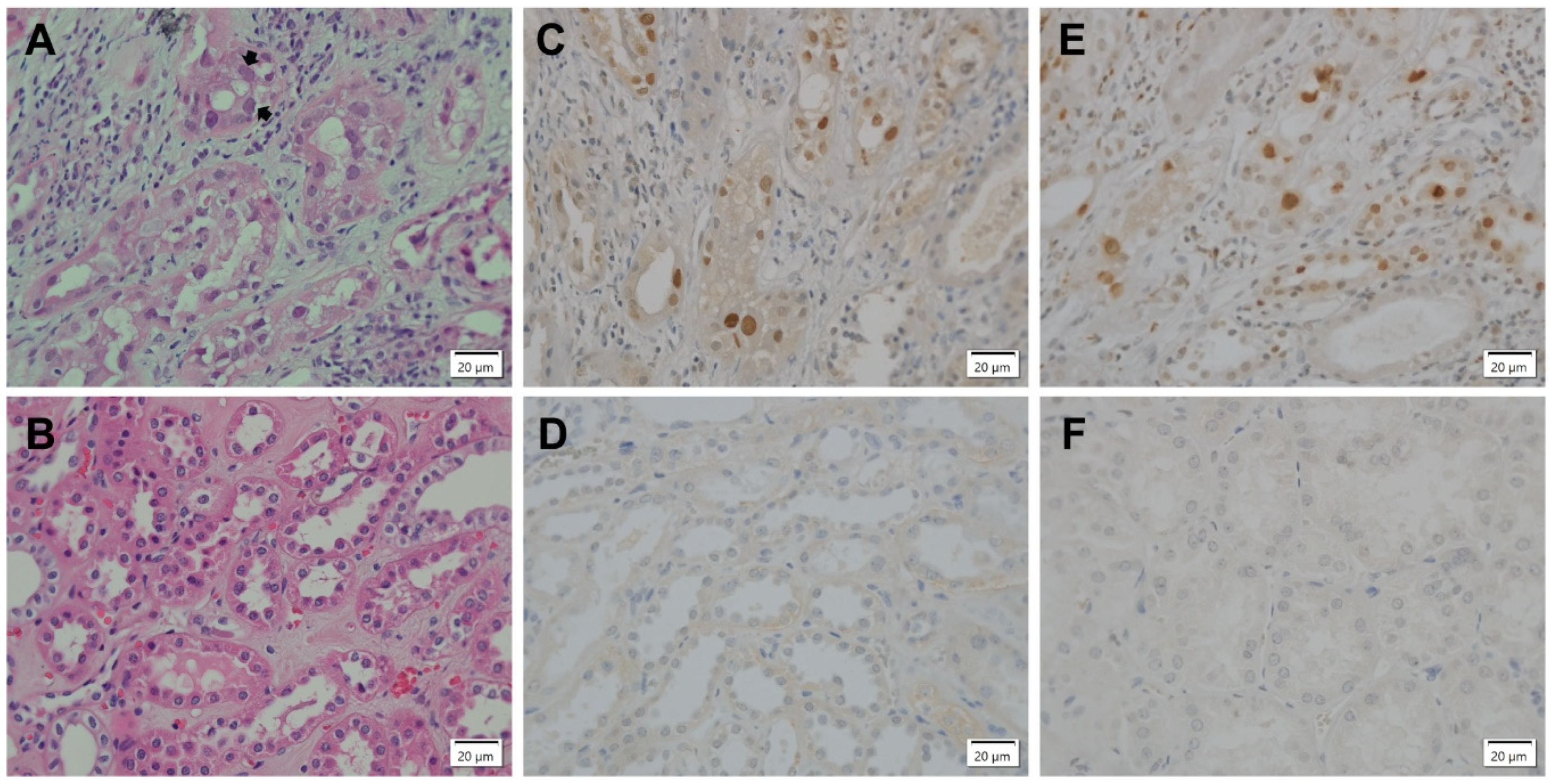
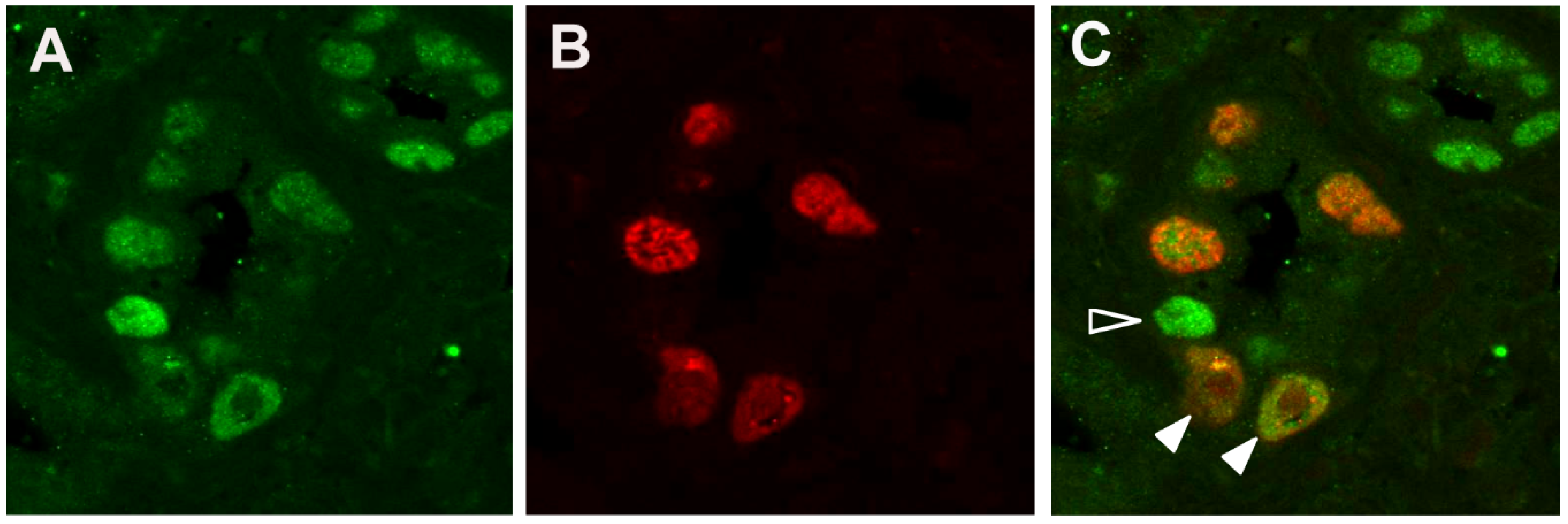
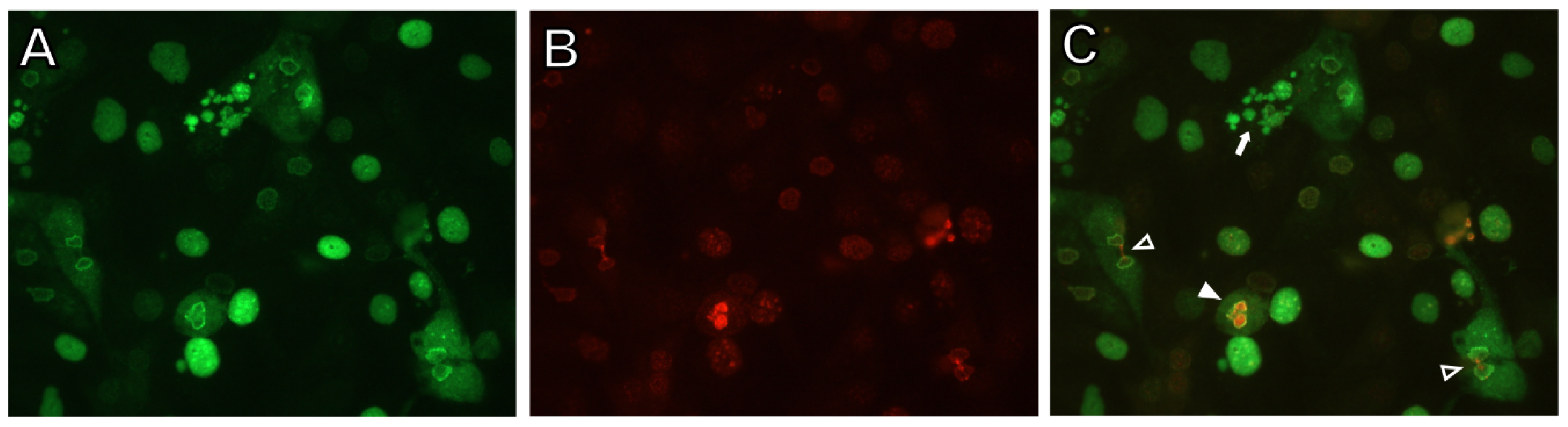
3.1. DDR Activation in BKPyV-Infected Cells in PyVAN
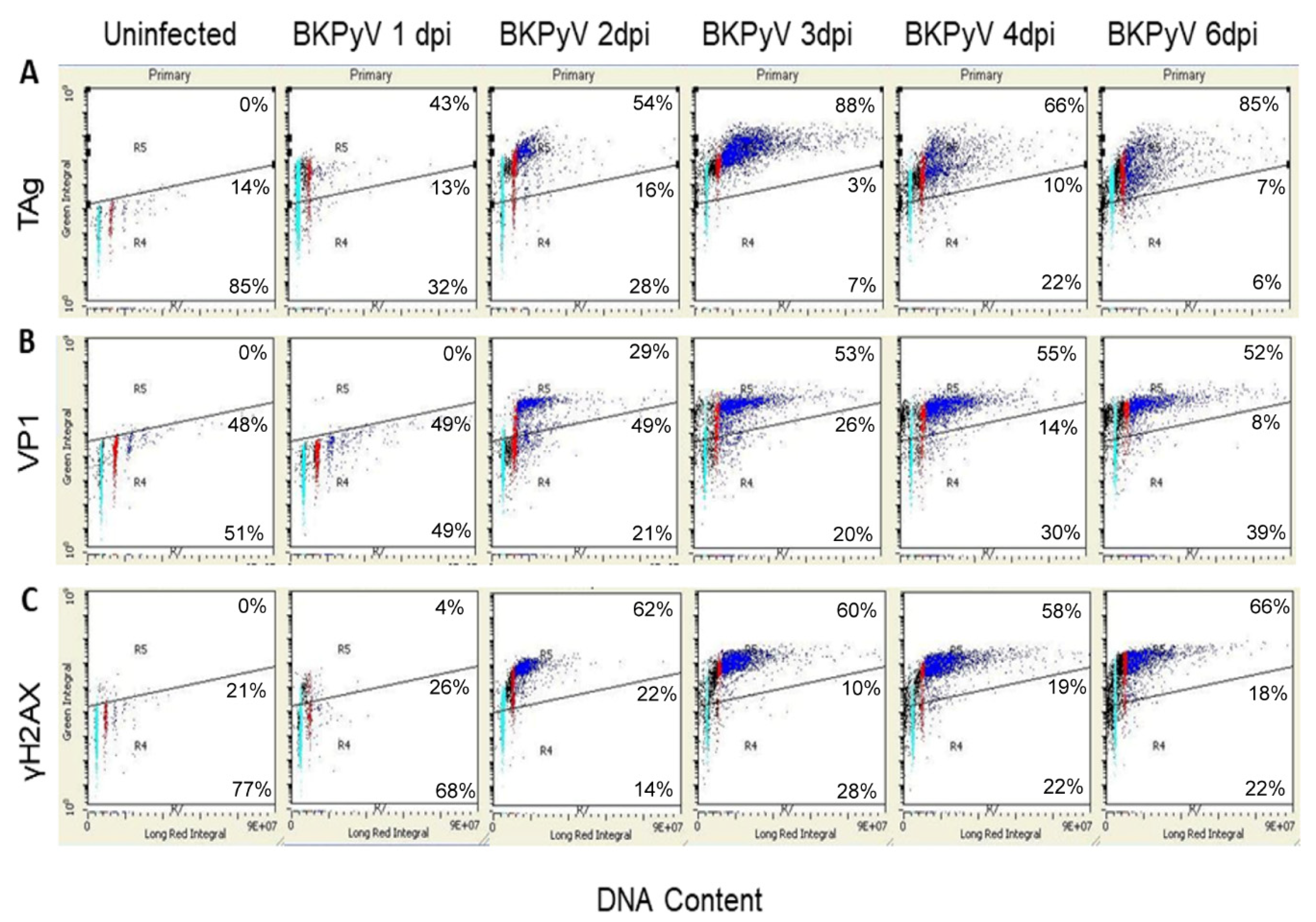
3.2. BKPyV Infection Stimulates DNA Synthesis beyond 4C in Cultured Human RPTECs
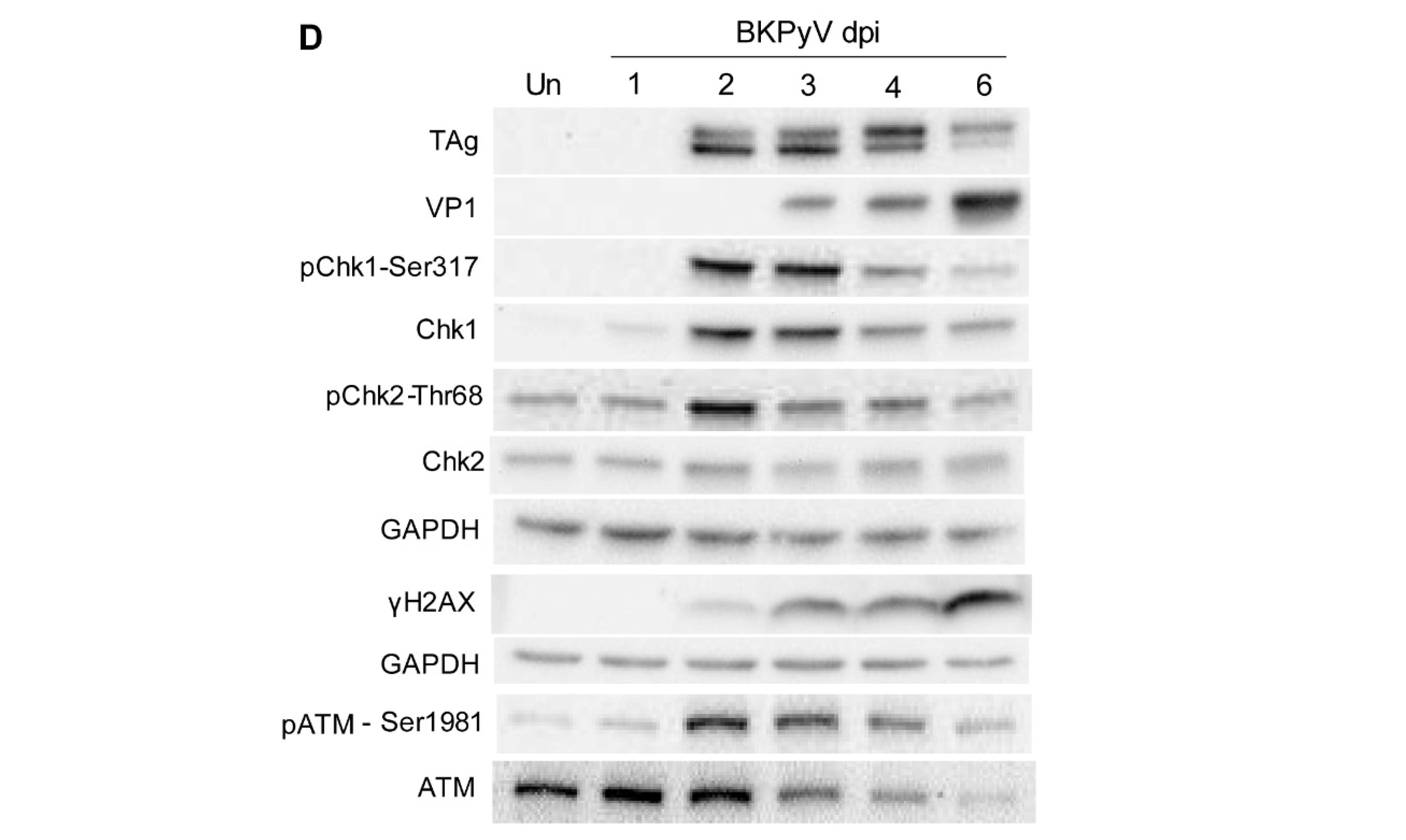
3.3. BKPyV Infection Stimulates a DDR in Cultured Human RPTECs
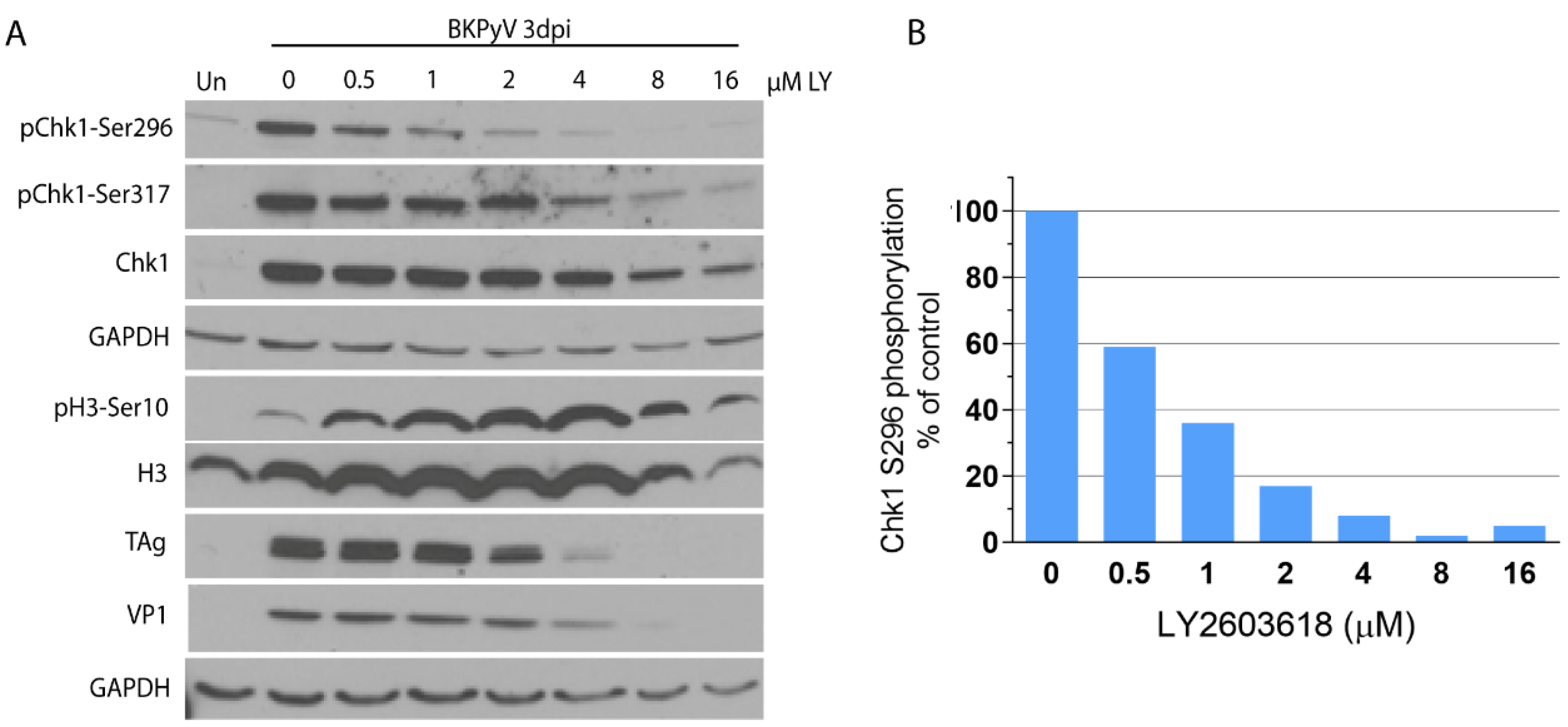
3.4. Inhibition of Chk1 by LY2603618 in BKPyV-Infected Cells
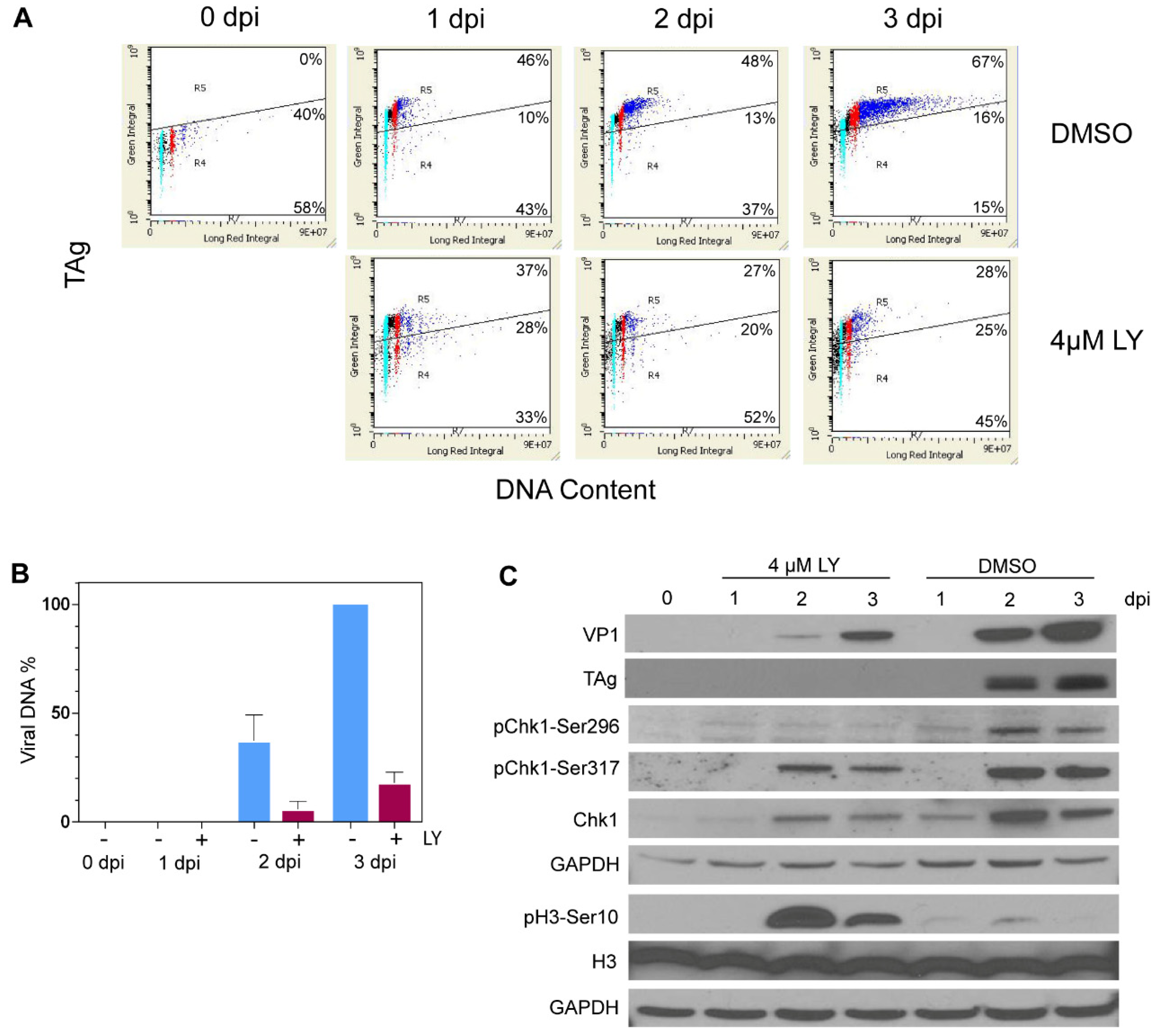
3.5. LY2603618 Restricts Progression of BKPyV-Infected Cells into >G2 Phase
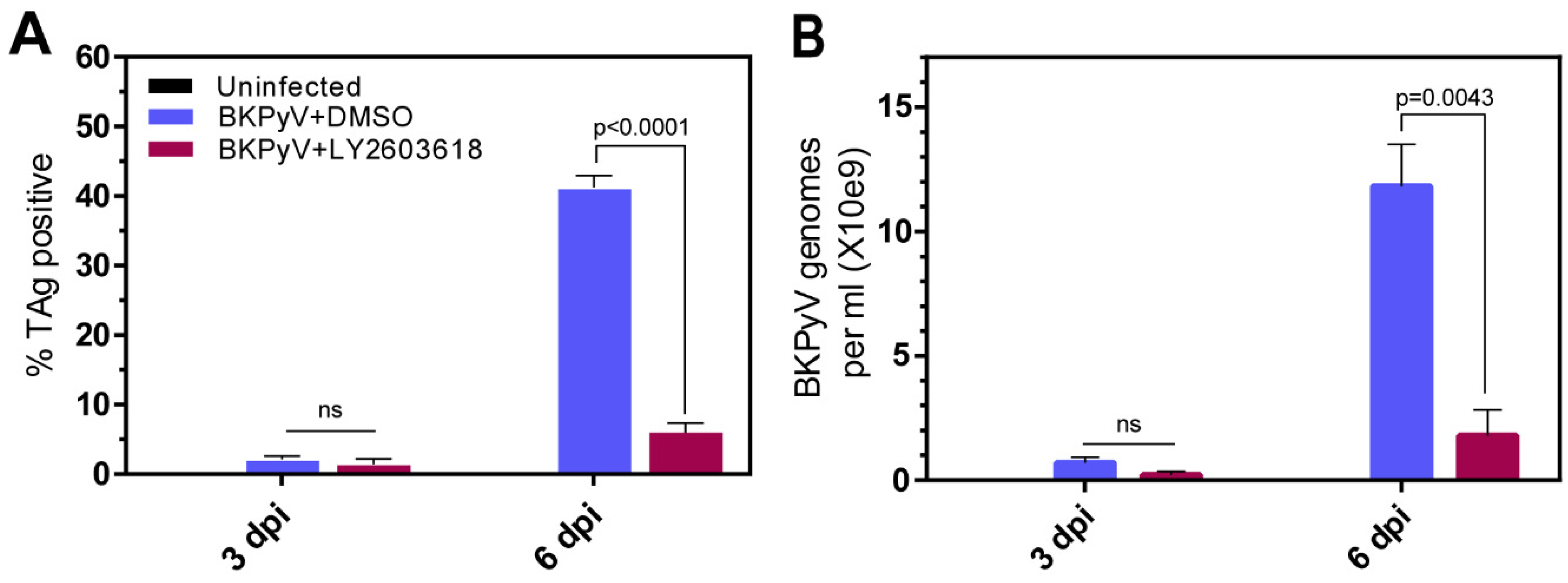
3.6. LY2603618 Reduces BKPyV Spread In Vitro
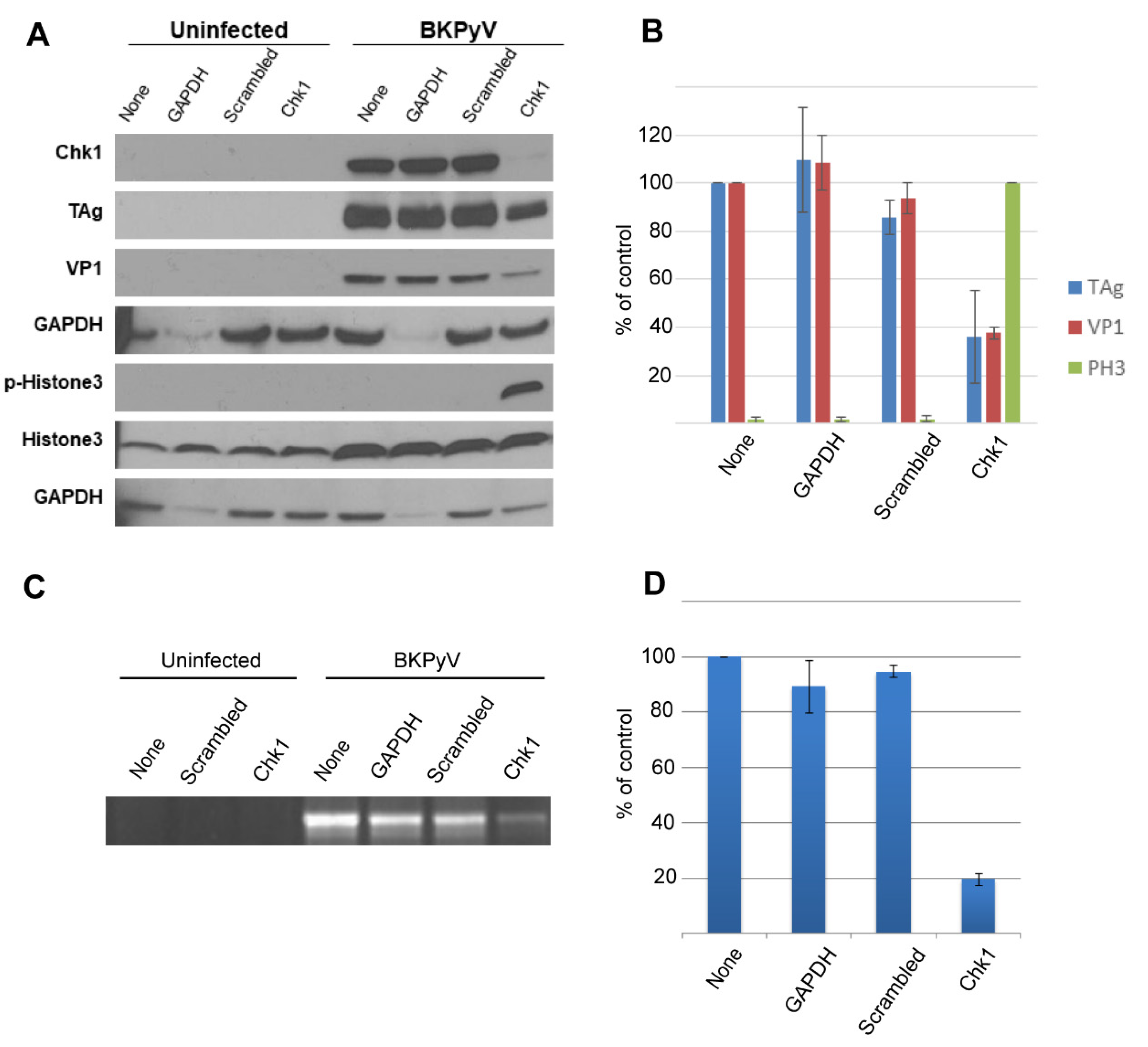
3.7. siRNA-Mediated Knockdown of Chk1 Inhibits Replication of BKPyV DNA
4. Discussion
4.1. BKPyV Infection of Human Renal Epithelium Induces a DNA Damage Response In Vivo and In Vitro
4.2. Chk1 Inhibition Restricts Continued >G2 Phase DNA Synthesis
4.3. Chk1 Inhibition and Stimulation of Mitotic Events
4.4. Chk1 Inhibition as an Antiviral Therapy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ambalathingal, G.R.; Francis, R.S.; Smyth, M.J.; Smith, C.; Khanna, R. BK Polyomavirus: Clinical Aspects, Immune Regulation, and Emerging Therapies. Clin. Microbiol. Rev. 2017, 30, 503–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS. Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imperiale, M.J.; Jiang, M. Polyomavirus Persistence. Annu. Rev. Virol. 2016, 3, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Kalvatchev, Z.; Slavov, S.; Shtereva, M.; Savova, S. Reactivation of Polyomavirus hominis 1 (BKV) during pregnancy and the risk of mother-to-child transmission1. J. Clin. Virol. 2008, 43, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Nickeleit, V.; Klimkait, T.; Binet, I.F.; Dalquen, P.; Del Zenero, V.; Thiel, G.; Mihatsch, M.J.; Hirsch, H.H. Testing for polyomavirus type BK DNA in plasma to identify renal-allograft recipients with viral nephropathy. N. Engl. J. Med. 2000, 342, 1309–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binet, I.; Nickeleit, V.; Hirsch, H.H.; Prince, O.; Dalquen, P.; Gudat, F.; Mihatsch, M.J.; Thiel, G. Polyomavirus disease under new immunosuppressive drugs: A cause of renal graft dysfunction and graft loss. Transplantation 1999, 67, 918–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randhawa, P.S.; Finkelstein, S.; Scantlebury, V.; Shapiro, R.; Vivas, C.; Jordan, M.; Picken, M.M.; Demetris, A.J. Human polyoma virus-associated interstitial nephritis in the allograft kidney. Transplantation 1999, 67, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Randhawa, P.S.; AST Infectious Diseases Community of Practice. BK polyomavirus in solid organ transplantation-Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13528. [Google Scholar] [CrossRef] [PubMed]
- Saade, A.; Styczynski, J.; Cesaro, S.; Infectious Disease Working party of EBMT. BK virus infection in allogeneic hematopoietic cell transplantation: An update on pathogenesis, immune responses, diagnosis and treatments. J. Infect. 2020, 81, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.S.; Gralla, J.; Chan, L.; Klem, P.; Wiseman, A.C. Aggressive immunosuppression minimization reduces graft loss following diagnosis of BK virus-associated nephropathy: A comparison of two reduction strategies. Clin. J. Am. Soc. Nephrol. 2008, 3, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Schaub, S.; Hirsch, H.H.; Dickenmann, M.; Steiger, J.; Mihatsch, M.J.; Hopfer, H.; Mayr, M. Reducing immunosuppression preserves allograft function in presumptive and definitive polyomavirus-associated nephropathy. Am. J. Transplant. 2010, 10, 2615–2623. [Google Scholar] [CrossRef]
- Petrov, R.; Elbahloul, O.; Gallichio, M.H.; Stellrecht, K.; Conti, D.J. Monthly screening for polyoma virus eliminates BK nephropathy and preserves renal function. Surg. Infect. 2009, 10, 85–90. [Google Scholar] [CrossRef]
- Saad, E.R.; Bresnahan, B.A.; Cohen, E.P.; Lu, N.; Orentas, R.J.; Vasudev, B.; Hariharan, S. Successful treatment of BK viremia using reduction in immunosuppression without antiviral therapy. Transplantation 2008, 85, 850–854. [Google Scholar] [CrossRef]
- Hardinger, K.L.; Koch, M.J.; Bohl, D.J.; Storch, G.A.; Brennan, D.C. BK-virus and the impact of pre-emptive immunosuppression reduction: 5-year results. Am. J. Transplant. 2010, 10, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Bischof, N.; Hirsch, H.H.; Wehmeier, C.; Amico, P.; Dickenmann, M.; Hirt-Minkowski, P.; Steiger, J.; Menter, T.; Helmut, H.; Schaub, S. Reducing calcineurin inhibitor first for treating BK polyomavirus replication after kidney transplantation: Long-term outcomes. Nephrol. Dial. Transplant. 2019, 34, 1240–1250. [Google Scholar] [CrossRef]
- Funk, G.A.; Steiger, J.; Hirsch, H.H. Rapid dynamics of polyomavirus type BK in renal transplant recipients. J. Infect. Dis. 2006, 193, 80–87. [Google Scholar] [CrossRef]
- Kadambi, P.V.; Josephson, M.A.; Williams, J.; Corey, L.; Jerome, K.R.; Meehan, S.M.; Limaye, A.P. Treatment of refractory BK virus-associated nephropathy with cidofovir. Am. J. Transplant. 2003, 3, 186–191. [Google Scholar] [CrossRef]
- Kuten, S.A.; Patel, S.J.; Knight, R.J.; Gaber, L.W.; DeVos, J.M.; Gaber, A.O. Observations on the use of cidofovir for BK virus infection in renal transplantation. Transpl. Infect. Dis. 2014, 16, 975–983. [Google Scholar] [CrossRef]
- Kuypers, D.R.; Vandooren, A.K.; Lerut, E.; Evenepoel, P.; Claes, K.; Snoeck, R.; Naesens, L.; Vanrenterghem, Y. Adjuvant low-dose cidofovir therapy for BK polyomavirus interstitial nephritis in renal transplant recipients. Am. J. Transplant. 2005, 5, 1997–2004. [Google Scholar] [CrossRef]
- Nesselhauf, N.; Strutt, J.; Bastani, B. Evaluation of leflunomide for the treatment of BK viremia and biopsy proven BK nephropathy; a single center experience. J. Nephropathol. 2016, 5, 34–37. [Google Scholar] [CrossRef]
- Josephson, M.A.; Gillen, D.; Javaid, B.; Kadambi, P.; Meehan, S.; Foster, P.; Harland, R.; Thistlethwaite, R.J.; Garfinkel, M.; Atwood, W.; et al. Treatment of renal allograft polyoma BK virus infection with leflunomide. Transplantation 2006, 81, 704–710. [Google Scholar] [CrossRef]
- Johnston, O.; Jaswal, D.; Gill, J.S.; Doucette, S.; Fergusson, D.A.; Knoll, G.A. Treatment of polyomavirus infection in kidney transplant recipients: A systematic review. Transplantation 2010, 89, 1057–1070. [Google Scholar] [CrossRef] [Green Version]
- Dropulic, L.K.; Cohen, J.I. Update on new antivirals under development for the treatment of double-stranded DNA virus infections. Clin. Pharmacol. Ther. 2010, 88, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Knoll, G.A.; Humar, A.; Fergusson, D.; Johnston, O.; House, A.A.; Kim, S.J.; Ramsay, T.; Chasse, M.; Pang, X.; Zaltzman, J.; et al. Levofloxacin for BK virus prophylaxis following kidney transplantation: A randomized clinical trial. JAMA 2014, 312, 2106–2114. [Google Scholar] [CrossRef]
- Shah, T.; Vu, D.; Naraghi, R.; Campbell, A.; Min, D. Efficacy of Intravenous Immunoglobulin in the Treatment of Persistent BK Viremia and BK Virus Nephropathy in Renal Transplant Recipients. Clin. Transpl. 2014, 109–116. Available online: https://pubmed.ncbi.nlm.nih.gov/26281134/.
- Sener, A.; House, A.A.; Jevnikar, A.M.; Boudville, N.; McAlister, V.C.; Muirhead, N.; Rehman, F.; Luke, P.P. Intravenous immunoglobulin as a treatment for BK virus associated nephropathy: One-year follow-up of renal allograft recipients. Transplantation 2006, 81, 117–120. [Google Scholar] [CrossRef]
- Hurdiss, D.L.; Morgan, E.L.; Thompson, R.F.; Prescott, E.L.; Panou, M.M.; Macdonald, A.; Ranson, N.A. New Structural Insights into the Genome and Minor Capsid Proteins of BK Polyomavirus using Cryo-Electron Microscopy. Structure 2016, 24, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Fink, L.H.; O’Hara, B.; Atwood, W.J.; Sullivan, C.S. Evolutionarily conserved function of a viral microRNA. J. Virol. 2008, 82, 9823–9828. [Google Scholar] [CrossRef] [Green Version]
- Manzetti, J.; Weissbach, F.H.; Graf, F.E.; Unterstab, G.; Wernli, M.; Hopfer, H.; Drachenberg, C.B.; Rinaldo, C.H.; Hirsch, H.H. BK Polyomavirus Evades Innate Immune Sensing by Disrupting the Mitochondrial Network and Promotes Mitophagy. Iscience 2020, 23, 101257. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, C.H.; Tylden, G.D.; Sharma, B.N. The human polyomavirus BK (BKPyV): Virological background and clinical implications. APMIS 2013, 121, 728–745. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Low, J.A.; Magnuson, B.; Tsai, B.; Imperiale, M.J. Identification of gangliosides GD1b and GT1b as receptors for BK virus. J. Virol. 2006, 80, 1361–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eash, S.; Querbes, W.; Atwood, W.J. Infection of vero cells by BK virus is dependent on caveolae. J. Virol. 2004, 78, 11583–11590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Marciano, A.T.; Rivet, C.R.; Imperiale, M.J. Caveolin- and clathrin-independent entry of BKPyV into primary human proximal tubule epithelial cells. Virology 2016, 492, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Abend, J.R.; Tsai, B.; Imperiale, M.J. Early events during BK virus entry and disassembly. J. Virol. 2009, 83, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Bennett, S.M.; Jiang, M.; Imperiale, M.J. Role of cell-type-specific endoplasmic reticulum-associated degradation in polyomavirus trafficking. J. Virol. 2013, 87, 8843–8852. [Google Scholar] [CrossRef] [Green Version]
- Bennett, S.M.; Zhao, L.; Bosard, C.; Imperiale, M.J. Role of a nuclear localization signal on the minor capsid proteins VP2 and VP3 in BKPyV nuclear entry. Virology 2015, 474, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Barth, H.; Solis, M.; Lepiller, Q.; Sueur, C.; Soulier, E.; Caillard, S.; Stoll-Keller, F.; Fafi-Kremer, S. 45 years after the discovery of human polyomaviruses BK and JC: Time to speed up the understanding of associated diseases and treatment approaches. Crit. Rev. Microbiol. 2017, 43, 178–195. [Google Scholar] [CrossRef]
- DeCaprio, J.; Imperiale, M.; Major, E. Polyomaviruses. In Fields Virology; Knipe, D., Howley, P., Eds.; Wolters Kluwer/Lippincot Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 1633–1661. [Google Scholar]
- Panou, M.M.; Antoni, M.; Morgan, E.L.; Loundras, E.A.; Wasson, C.W.; Welberry-Smith, M.; Mankouri, J.; Macdonald, A. Glibenclamide inhibits BK polyomavirus infection in kidney cells through CFTR blockade. Antivir. Res. 2020, 178, 104778. [Google Scholar] [CrossRef]
- Kane, J.R.; Fong, S.; Shaul, J.; Frommlet, A.; Frank, A.O.; Knapp, M.; Bussiere, D.E.; Kim, P.; Ornelas, E.; Cuellar, C.; et al. A polyomavirus peptide binds to the capsid VP1 pore and has potent antiviral activity against BK and JC polyomaviruses. Elife 2020, 9, e50722. [Google Scholar] [CrossRef]
- Dahl, J.; You, J.; Benjamin, T.L. Induction and utilization of an ATM signaling pathway by polyomavirus. J. Virol. 2005, 79, 13007–13017. [Google Scholar] [CrossRef] [Green Version]
- Orba, Y.; Suzuki, T.; Makino, Y.; Kubota, K.; Tanaka, S.; Kimura, T.; Sawa, H. Large T antigen promotes JC virus replication in G2-arrested cells by inducing ATM- and ATR-mediated G2 checkpoint signaling. J. Biol. Chem. 2010, 285, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Madden-Fuentes, R.J.; Lou, B.X.; Pipas, J.M.; Gerhardt, J.; Rigell, C.J.; Fanning, E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J. Virol. 2008, 82, 5316–5328. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Dodson, G.E.; Shaikh, S.; Rundell, K.; Tibbetts, R.S. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 2005, 280, 40195–40200. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhao, L.; Gamez, M.; Imperiale, M.J. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS. Pathog. 2012, 8, e1002898. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Darbinyan, A.; White, M.K.; Akan, S.; Radhakrishnan, S.; Del, V.L.; Amini, S.; Khalili, K. Alterations of DNA damage repair pathways resulting from JCV infection. Virology 2007, 364, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Wang, X.; Li, J.; Buck, C.B.; You, J. Host DNA damage response factors localize to merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J. Virol. 2014, 88, 3285–3297. [Google Scholar] [CrossRef] [Green Version]
- Heiser, K.; Nicholas, C.; Garcea, R.L. Activation of DNA damage repair pathways by murine polyomavirus. Virology 2016, 497, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Lou, S.; Luo, Y.; Cheng, F.; Huang, Q.; Shen, W.; Kleiboeker, S.; Tisdale, J.F.; Liu, Z.; Qiu, J. Human parvovirus B19 DNA replication induces a DNA damage response that is dispensable for cell cycle arrest at phase G2/M. J. Virol. 2012, 86, 10748–10758. [Google Scholar] [CrossRef] [Green Version]
- Jha, H.C.; Upadhyay, S.K.; AJ, M.P.; Lu, J.; Cai, Q.; Saha, A.; Robertson, E.S. H2AX phosphorylation is important for LANA-mediated Kaposi’s sarcoma-associated herpesvirus episome persistence. J. Virol. 2013, 87, 5255–5269. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.V.; Dutta, D.; Ansari, M.A.; Dutta, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J. Virol. 2014, 88, 2821–2834. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, R.; Skalka, G.L.; Stewart, G.S.; Hislop, A.D.; Blackbourn, D.J.; Grand, R.J. Activation of DNA Damage Response Pathways during Lytic Replication of KSHV. Viruses 2015, 7, 2908–2927. [Google Scholar] [CrossRef]
- Botting, C.; Lu, X.; Triezenberg, S.J. H2AX phosphorylation and DNA damage kinase activity are dispensable for herpes simplex virus replication. Virol. J. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- E, X.; Pickering, M.T.; Debatis, M.; Castillo, J.; Lagadinos, A.; Wang, S.; Lu, S.; Kowalik, T.F. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS. Pathog. 2011, 7, e1001342. [Google Scholar] [CrossRef]
- Nichols, G.J.; Schaack, J.; Ornelles, D.A. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J. Virol. 2009, 83, 5987–5998. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Hou, N.B.; Song, T.; He, X.; Zheng, Z.R.; Ma, Q.J.; Li, L.; Zhang, Y.H.; Zhong, H. Cellular DNA repair cofactors affecting hepatitis B virus infection and replication. World J. Gastroenterol. 2008, 14, 5059–5065. [Google Scholar] [CrossRef]
- Justice, J.L.; Needham, J.M.; Thompson, S.R. BK Polyomavirus Activates the DNA Damage Response to Prolong S Phase. J. Virol. 2019, 93, e00130-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, E.; Braiteh, F.; Von Hoff, D.; McWilliams, R.; Becerra, C.; Galsky, M.D.; Jameson, G.; Lin, J.; McKane, S.; Wickremsinhe, E.R.; et al. Phase I Study of CHK1 Inhibitor LY2603618 in Combination with Gemcitabine in Patients with Solid Tumors. Oncology 2016, 91, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Chen, V.J.; Marshall, M.; Ohnmacht, U.; Hynes, S.M.; Kumm, E.; Diaz, H.B.; Barnard, D.; Merzoug, F.F.; Huber, L.; et al. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Investig. New Drugs 2014, 32, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Laquente, B.; Lopez-Martin, J.; Richards, D.; Illerhaus, G.; Chang, D.Z.; Kim, G.; Stella, P.; Richel, D.; Szcylik, C.; Cascinu, S.; et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 2017, 17, 137. [Google Scholar] [CrossRef] [Green Version]
- Wehler, T.; Thomas, M.; Schumann, C.; Bosch-Barrera, J.; Vinolas Segarra, N.; Dickgreber, N.J.; Dalhoff, K.; Sebastian, M.; Corral Jaime, J.; Alonso, M.; et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017, 108, 212–216. [Google Scholar] [CrossRef]
- Doi, T.; Yoshino, T.; Shitara, K.; Matsubara, N.; Fuse, N.; Naito, Y.; Uenaka, K.; Nakamura, T.; Hynes, S.M.; Lin, A.B. Phase I study of LY2603618, a CHK1 inhibitor, in combination with gemcitabine in Japanese patients with solid tumors. Anticancer Drugs 2015, 26, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.; Kang, J.H.; Smith, D.; Rosenberg, R.; Park, K.; Kim, S.W.; Su, W.C.; Boyd, T.E.; Richards, D.A.; Novello, S.; et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Investig. New Drugs 2016, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Hynes, S.M.; Wickremsinhe, E.; Zhang, W.; Decker, R.; Ott, J.; Chandler, J.; Mitchell, M. Evaluation of the likelihood of a selective CHK1 inhibitor (LY2603618) to inhibit CYP2D6 with desipramine as a probe substrate in cancer patients. Biopharm. Drug Dispos. 2015, 36, 49–63. [Google Scholar] [CrossRef]
- Abend, J.R.; Low, J.A.; Imperiale, M.J. Inhibitory effect of gamma interferon on BK virus gene expression and replication. J. Virol. 2007, 81, 272–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, A.T.; Wolfe, D. Tissue processing and hematoxylin and eosin staining. Methods Mol. Biol. 2014, 1180, 31–43. [Google Scholar]
- Friedrich, T.D.; Smith, R.L.; Lehman, J.M. Quantitation of Cellular Proteins by Laser Scanning Cytometry. In The Protein Protocols Handbook, 3rd ed.; Walker, J.W., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 63–69. [Google Scholar]
- Yeung, Y.G.; Stanley, E.R. A solution for stripping antibodies from polyvinylidene fluoride immunoblots for multiple reprobing. Anal. Biochem. 2009, 389, 89–91. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, K.; Bui, T.; Frisque, R.J.; Grandinetti, A.; Nerurkar, V.R. A rapid in vitro polyomavirus DNA replication assay. J. Virol. Methods 2004, 122, 123–127. [Google Scholar] [CrossRef]
- Dumoulin, A.; Hirsch, H.H. Reevaluating and optimizing polyomavirus BK and JC real-time PCR assays to detect rare sequence polymorphisms. J. Clin. Microbiol. 2011, 49, 1382–1388. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, H.H.; Brennan, D.C.; Drachenberg, C.B.; Ginevri, F.; Gordon, J.; Limaye, A.P.; Mihatsch, M.J.; Nickeleit, V.; Ramos, E.; Randhawa, P.; et al. Polyomavirus-associated nephropathy in renal transplantation: Interdisciplinary analyses and recommendations. Transplantation 2005, 79, 1277–1286. [Google Scholar] [CrossRef]
- Drachenberg, C.B.; Hirsch, H.H.; Ramos, E.; Papadimitriou, J.C. Polyomavirus disease in renal transplantation: Review of pathological findings and diagnostic methods. Hum. Pathol. 2005, 36, 1245–1255. [Google Scholar] [CrossRef]
- Drachenberg, C.B.; Papadimitriou, J.C.; Hirsch, H.H.; Wali, R.; Crowder, C.; Nogueira, J.; Cangro, C.B.; Mendley, S.; Mian, A.; Ramos, E. Histological patterns of polyomavirus nephropathy: Correlation with graft outcome and viral load. Am. J. Transplant. 2004, 4, 2082–2092. [Google Scholar] [CrossRef]
- Seemayer, C.A.; Seemayer, N.H.; Durmuller, U.; Gudat, F.; Schaub, S.; Hirsch, H.H.; Mihatsch, M.J. BK virus large T and VP-1 expression in infected human renal allografts. Nephrol. Dial. Transplant. 2008, 23, 3752–3761. [Google Scholar] [CrossRef] [Green Version]
- Wiley, S.R.; Kraus, R.J.; Zuo, F.; Murray, E.E.; Loritz, K.; Mertz, J.E. SV40 early-to-late switch involves titration of cellular transcriptional repressors. Genes Dev. 1993, 7, 2206–2219. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Donehower, R.C.; Iyengar, T.; Ramanathan, R.K.; Lewandowski, K.; Westin, E.; Hurt, K.; Hynes, S.M.; Anthony, S.P.; McKane, S. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Investig. New Drugs 2013, 31, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.; Diaz, H.; Barnard, D.; Barda, D.; Clawson, D.; Blosser, W.; Cox, K.; Guo, S.; Marshall, M. Characterization and preclinical development of LY2603618: A selective and potent Chk1 inhibitor. Investig. New Drugs 2014, 32, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.L.; Verhalen, B.; Jiang, M. Polyomavirus interaction with the DNA damage response. Virol. Sin. 2015, 30, 122–129. [Google Scholar] [CrossRef]
- Weinreb, D.B.; Desman, G.T.; Burstein, D.E.; Kim, D.U.; Dikman, S.H.; Johnson, E.M. Expression of p53 in virally infected tubular cells in renal transplant patients with polyomavirus nephropathy. Hum. Pathol. 2006, 37, 684–688. [Google Scholar] [CrossRef]
- Wiesend, W.N.; Parasuraman, R.; Li, W.; Farinola, M.A.; Rooney, M.T.; Hick, S.K.; Samarapungavan, D.; Cohn, S.R.; Reddy, G.H.; Rocher, L.L.; et al. Adjuvant role of p53 immunostaining in detecting BK viral infection in renal allograft biopsies. Ann. Clin. Lab. Sci. 2010, 40, 324–329. [Google Scholar]
- Hu, L.; Filippakis, H.; Huang, H.; Yen, T.J.; Gjoerup, O.V. Replication stress and mitotic dysfunction in cells expressing simian virus 40 large T antigen. J. Virol. 2013, 87, 13179–13192. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.M.; Laffin, J.; Jacobberger, J.W.; Fogleman, D. Analysis of simian virus 40 infection of CV-1 cells by quantitative two-color fluorescence with flow cytometry. Cytometry 1988, 9, 52–59. [Google Scholar] [CrossRef]
- Lehman, J.M.; Laffin, J.; Friedrich, T.D. Flow cytometry of DNA increase after simian virus 40 infection of CV-1 cells. In Vitro Cell. Dev. Biol. 1992, 28, 306–308. [Google Scholar]
- Friedrich, T.D.; Laffin, J.; Lehman, J.M. Simian virus 40 large T-antigen function is required for induction of tetraploid DNA content during lytic infection. J. Virol. 1992, 66, 4576–4579. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.M.; Laffin, J.; Friedrich, T.D. Simian virus 40 induces multiple S phases with the majority of viral DNA replication in the G2 and second S phase in CV-1 cells. Exp. Cell Res. 2000, 258, 215–222. [Google Scholar] [CrossRef]
- Verhalen, B.; Justice, J.L.; Imperiale, M.J.; Jiang, M. Viral DNA replication-dependent DNA damage response activation during BK polyomavirus infection. J. Virol. 2015, 89, 5032–5039. [Google Scholar] [CrossRef] [Green Version]
- Jul-Larsen, A.; Visted, T.; Karlsen, B.O.; Rinaldo, C.H.; Bjerkvig, R.; Lonning, P.E.; Boe, S.O. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp. Cell Res. 2004, 298, 58–73. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar]
- Zhu, W.; Dutta, A. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol. Cell. Biol. 2006, 26, 4601–4611. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Seiler, J.A.; Burhans, W.C. Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J. Biol. Chem. 2003, 278, 4295–4304. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, Y.S.; Watanabe, N.; Morisaki, H.; Akita, H.; Fujimoto, A.; Tominaga, K.; Terasawa, M.; Tachibana, A.; Ikeda, K.; Nakanishi, M. Cell-cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene 1999, 18, 3673–3681. [Google Scholar]
- Carrassa, L.; Broggini, M.; Vikhanskaya, F.; Damia, G. Characterization of the 5′flanking region of the human Chk1 gene: Identification of E2F1 functional sites. Cell Cycle 2003, 2, 604–609. [Google Scholar]
- White, M.K.; Khalili, K. Interaction of retinoblastoma protein family members with large T-antigen of primate polyomaviruses. Oncogene 2006, 25, 5286–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.W.; Otterness, D.M.; Chiang, G.G.; Xie, W.; Liu, Y.C.; Mercurio, F.; Abraham, R.T. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 2005, 19, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Leung-Pineda, V.; Huh, J.; Piwnica-Worms, H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 2009, 69, 2630–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.W.; Brognard, J.; Coughlin, C.; You, Z.; Dolled-Filhart, M.; Aslanian, A.; Manning, G.; Abraham, R.T.; Hunter, T. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol. Cell 2009, 35, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, K.B.; Bang, S.; Kurokawa, M.; Gerber, S.A. Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. FEBS J. 2020, 287, 1985–1999. [Google Scholar] [CrossRef]
- Kim, K.S.; Heo, J.I.; Choi, K.J.; Bae, S. Enhancement of cellular radiation sensitivity through degradation of Chk1 by the XIAP-XAF1 complex. Cancer Biol. Ther. 2014, 15, 1622–1634. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Liu, H.; Zhu, X.; Shen, H.; Ma, X.; Wang, F.; Huang, M.; Gong, J.; Li, X.; Wang, Y.; et al. Ataxin-3 promotes genome integrity by stabilizing Chk1. Nucleic Acids Res. 2017, 45, 4532–4549. [Google Scholar] [CrossRef] [Green Version]
- Alvarez Orellana, J.; Kwun, H.J.; Artusi, S.; Chang, Y.; Moore, P.S. Sirolimus and other mTOR inhibitors directly activate latent pathogenic human polyomavirus replication. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Brooks, K.; Oakes, V.; Edwards, B.; Ranall, M.; Leo, P.; Pavey, S.; Pinder, A.; Beamish, H.; Mukhopadhyay, P.; Lambie, D.; et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene 2013, 32, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Zachos, G.; Rainey, M.D.; Gillespie, D.A. Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol. 2005, 25, 563–574. [Google Scholar] [CrossRef] [Green Version]
- McNeely, S.; Conti, C.; Sheikh, T.; Patel, H.; Zabludoff, S.; Pommier, Y.; Schwartz, G.; Tse, A. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle 2010, 9, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Okubo, E.; Lehman, J.M.; Friedrich, T.D. Negative regulation of mitotic promoting factor by the checkpoint kinase chk1 in simian virus 40 lytic infection. J. Virol. 2003, 77, 1257–1267. [Google Scholar] [CrossRef] [Green Version]
- Scarano, F.J.; Laffin, J.A.; Lehman, J.M.; Friedrich, T.D. Simian virus 40 prevents activation of M-phase-promoting factor during lytic infection. J. Virol. 1994, 68, 2355–2361. [Google Scholar] [CrossRef] [Green Version]
- Acott, P.; O’Regan, P.A.; Crocker, J.F. Suppression of early and chronic BK polyoma virus replication by mycophenolic acid in Vero cells. Transpl. Int. 2009, 22, 225–231. [Google Scholar] [CrossRef]
- Warren, N.J.H.; Eastman, A. Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase I inhibitors when used as single agents or in combination with DNA damage. Oncogene 2020, 39, 1389–1401. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hainley, L.E.; Hughson, M.S.; Narendran, A.; Smith, R.; Arthur, J.; Hayner-Buchan, A.; Conti, D.J.; Lehman, J.M.; Friedrich, T.D. Chk1 and the Host Cell DNA Damage Response as a Potential Antiviral Target in BK Polyomavirus Infection. Viruses 2021, 13, 1353. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071353
Hainley LE, Hughson MS, Narendran A, Smith R, Arthur J, Hayner-Buchan A, Conti DJ, Lehman JM, Friedrich TD. Chk1 and the Host Cell DNA Damage Response as a Potential Antiviral Target in BK Polyomavirus Infection. Viruses. 2021; 13(7):1353. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071353
Chicago/Turabian StyleHainley, Lydia E., Martina S. Hughson, Amithi Narendran, Ralph Smith, Justin Arthur, Alida Hayner-Buchan, David J. Conti, John M. Lehman, and Thomas D. Friedrich. 2021. "Chk1 and the Host Cell DNA Damage Response as a Potential Antiviral Target in BK Polyomavirus Infection" Viruses 13, no. 7: 1353. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071353