
Precursors of Viral Proteases as Distinct Drug Targets


1
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Prague, Czech Republic
2
Department of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, 128 43 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Viruses 2021, 13(10), 1981; https://0-doi-org.brum.beds.ac.uk/10.3390/v13101981
Submission received: 31 August 2021
/
Revised: 25 September 2021
/
Accepted: 28 September 2021
/
Published: 2 October 2021
(This article belongs to the Special Issue Virology in the Czech Republic – from a Great Legacy to Optimistic Future)
Abstract
:Viral proteases are indispensable for successful virion maturation, thus making them a prominent drug target. Their enzyme activity is tightly spatiotemporally regulated by expression in the precursor form with little or no activity, followed by activation via autoprocessing. These cleavage events are frequently triggered upon transportation to a specific compartment inside the host cell. Typically, precursor oligomerization or the presence of a co-factor is needed for activation. A detailed understanding of these mechanisms will allow ligands with non-canonical mechanisms of action to be designed, which would specifically modulate the initial irreversible steps of viral protease autoactivation. Binding sites exclusive to the precursor, including binding sites beyond the protease domain, can be exploited. Both inhibition and up-regulation of the proteolytic activity of viral proteases can be detrimental for the virus. All these possibilities are discussed using examples of medically relevant viruses including herpesviruses, adenoviruses, retroviruses, picornaviruses, caliciviruses, togaviruses, flaviviruses, and coronaviruses.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Precursors as Major Signalization and Orchestration Agents
Autoproteolytic processing of protease precursors releases the active enzyme and the adjacent sequence(s). These released peptides or proteins can have other specific functions. The earliest proteins discovered to be autoactivated are digestive enzymes such as pepsin, trypsin, or chymotrypsin [1,2,3,4,5,6,7]. The blood clotting cascade is an example of regulation via proteolytic autoactivation in the human body [8]. Inherent activity not only of the mature enzyme but also of the precursor form was first reviewed 50 years ago [1]. The first self-processing and self-activating enzymes were observed in 1966 [9].
Proteases often act as a trigger-point of biological processes and their activation via autoprocessing is irreversible. Not surprisingly, the self-cleavage of the precursor is tightly spatiotemporally regulated in order to occur in the correct location with optimal rates. In many cases, details of these processes remain unknown. Compartmentalization often plays a role in autoactivation, as exemplified by the lysosomal protease cathepsin D. The migration of procathepsin D to the lysosome leads to the release of the active protease from the precursor proenzyme due to the acidic pH of this compartment [10,11,12,13]. Autoprocessing also functions in signaling pathways when an N-terminal or C-terminal-signaling molecule is released from the precursor after self-cleavage. If a signaling molecule is present at both terminals, it can be involved in two independent regulatory pathways [14]. The propart (usually the sequence adjacent to the N-terminus of a protease) can have a intramolecular chaperone function and can guide proper enzyme folding [15].
Viral Polyprotein Strategy
Viruses use various strategies for protein expression. One of these is to synthesize viral proteins, including one or more proteases, in the form of a polyprotein. Viral proteases are capable of autocatalytic release, along with cleavage of the viral polyprotein into separate functional proteins during the maturation process. Protein complex formation, timely differentiated steric accessibility of cleavage sites, and the interaction with host cell membrane organelles are all often involved in the regulation of proteolysis during viral maturation [16,17,18,19].
The polyprotein strategy is employed by positive RNA viruses, including retroviruses such as HIV (Human Immunodeficiency Virus) [17]; coronaviruses such as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [20]; and by some DNA viruses, e.g., poxviruses (smallpox virus) [21] or herpesviruses [22]. Other DNA viruses, such as polyomaviruses [23,24], all negative RNA viruses (among them, influenza virus [25]), and double-stranded RNA viruses [26] use different strategies of protein expression and encode no protease.
Inhibition of a specific viral protease interrupts the viral life cycle by abolishing the production of viral proteins needed for the replication and spread of the virus. Active-site inhibitors of HIV and Hepatitis C Virus (HCV) proteases are in clinical use [17,27]. Compounds targeting viral proteases through a mechanism other than the inhibition of the active site could broaden the portfolio of antivirals to overcome the development of drug resistance and to solve problems regarding non-specificity, toxicity, or improper pharmacokinetic properties. The multifunctional nature of viral protease precursors offers several potential ways for antiviral therapeutic interventions. The strategies are discussed below.
2. DNA Viruses
2.1. Herpesviruses
Herpesviruses are enveloped DNA viruses causing diverse human diseases, such as cold sores or chicken pox. Some herpesviruses can establish persistent infections and are also important opportunistic pathogens. Several nucleoside derivatives targeting the viral replication machinery are in clinical use [28,29,30]. Efforts to develop inhibitors of the herpes protease-assemblinhave not yet yielded any clinical candidates.
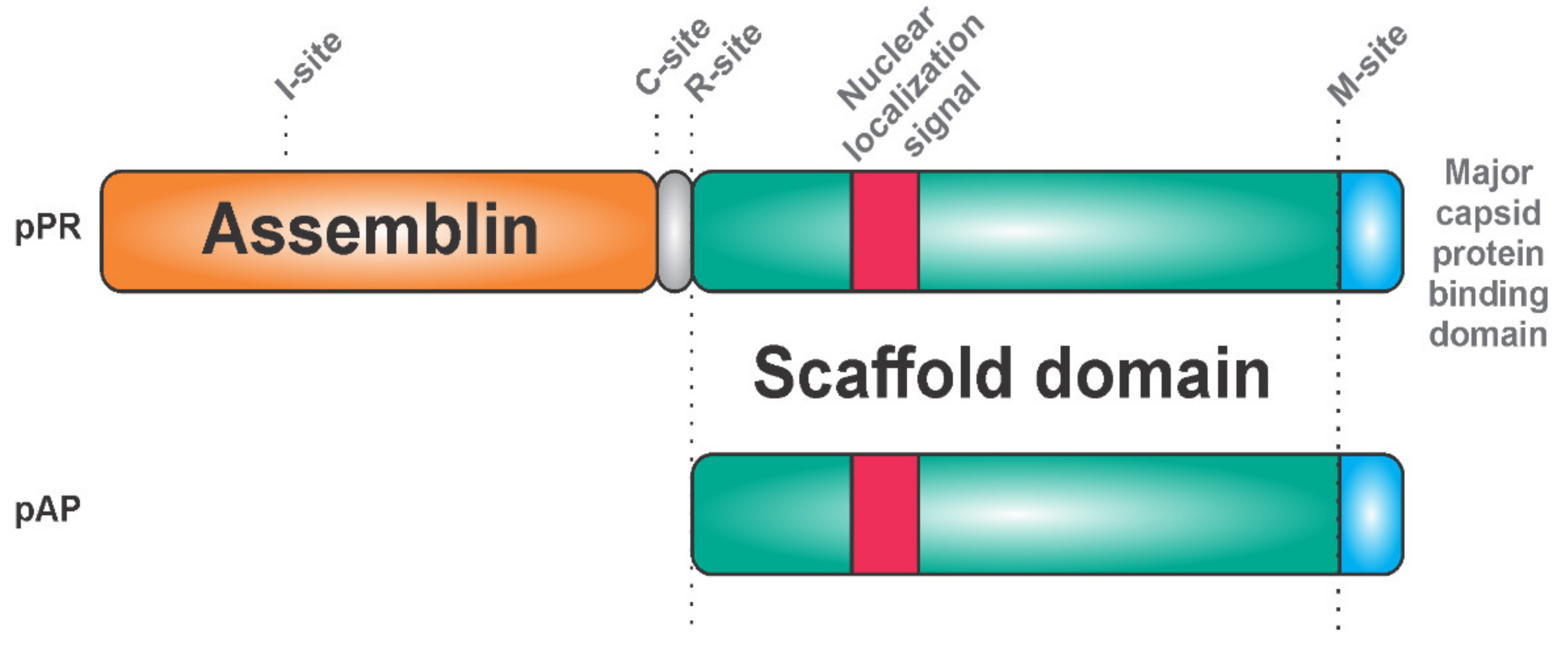
Herpesviruses harbor a serine protease, which has a unique catalytic Ser-His-His triad and is active as a homodimer [31,32,33,34]. The protease is C-terminally fused with a scaffold protein, which has a major capsid protein-binding motif on its C-terminus [35,36] and a nuclear localization signal on its N-terminus (Figure 1).
The protease precursor pPR (expressed from the gene denoted as UL26) binds to the major capsid protein (this interaction probably inhibits its proteolytic activity) and results in the transportation of the whole complex into the cell nucleus, the site of virion assembly. In the nucleus, the scaffold domains of the protease precursor proteins self-associate into spherical procapsids, in which the protease domains come into its proximity [37]. Protease dimerization leads to conformational rearrangements, which result in a dramatic increase in proteolytic activity. Cleavage at two sites of the precursor (denoted as M and R) releases the scaffold protein. Detailed studies of the human cytomegalovirus (HCMV) also revealed two intra-assemblin cleavage sites [38]. Cleavage at the C site leads to assemblin inactivation, whereas processing at the I site generates non-covalently associated fragments of assemblin with retained activity [39]. Cleavage at the I site triggers structural rearrangements, which bury the C site and prevent its hydrolysis. Inhibiting hydrolysis at the C and I site by mutagenesis synergistically reduces virus infectivity by 90% [40]. It seems that the precise equilibrium between the active and inactive forms of assemblin is important for determining whether virus replication and spreading is successful.
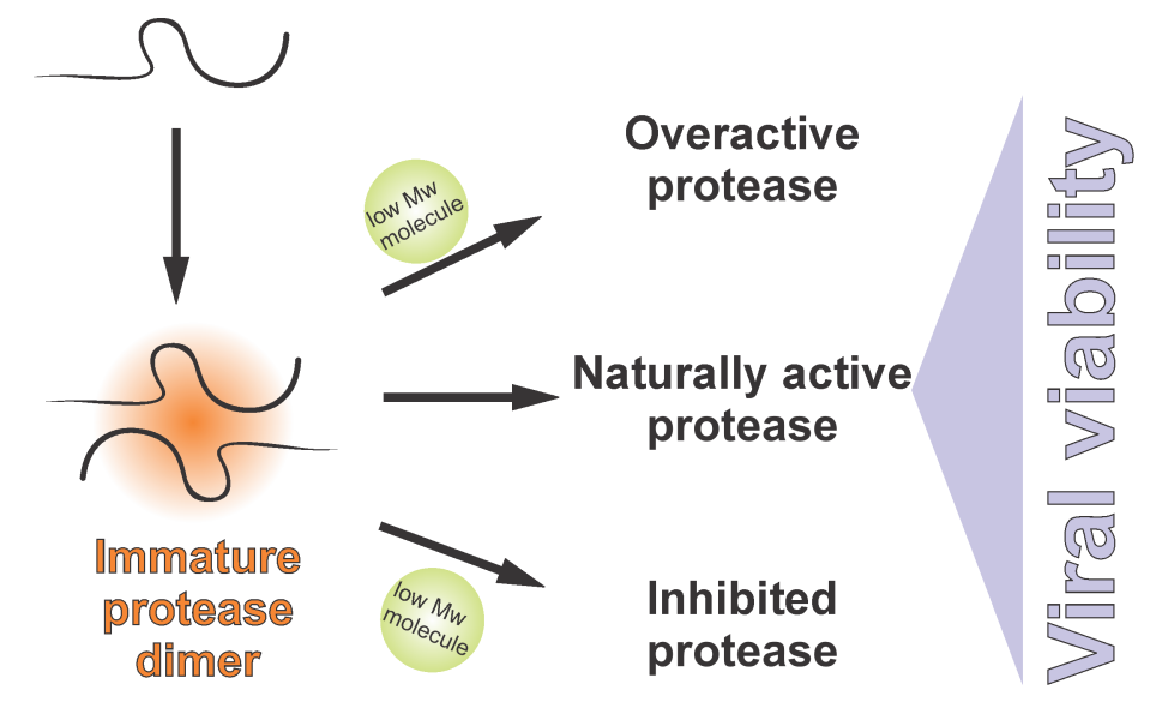
Since the herpes virus protease is active in its dimeric form, disruption of inter-monomeric protein–protein interactions represents another option to design inhibitors. Peptides and alpha-helix mimetics were reported as inhibitors disrupting the dimerization interfaces between the two monomers [41,42,43]. Dimerization inhibitors might also act at the level of the pPR precursor. Compounds blocking protein–protein interactions of the pPR precursor in the scaffold domain could represent an interesting alternative for drug design. Even more speculative is the possibility of exploiting the scaffold domain for artificial premature activation of assemblin. Up-regulation of the proteolytic activity could be as detrimental for the virus as its down-regulation by classical inhibitors.
Experiments with purified enzymes showed that the protease can be overactivated. Specifically, artificial activation by cosmotropic agents, such as ammonium sulphate, increases the activity of assemblin by several orders of magnitude [44].
Another homologous herpesvirus protease with deubiquitinating activity was identified in a conserved area of the Herpes simplex virus-1 genome. This protease is embedded within the N-terminal, part of the large tegument protein UL36, and is active only after proteolytic release from its precursor [45,46,47]. It seems to be involved in the viral replication [48] and modulation of host cell antiviral responses [49,50], making it another viable drug target.
2.2. Adenoviruses
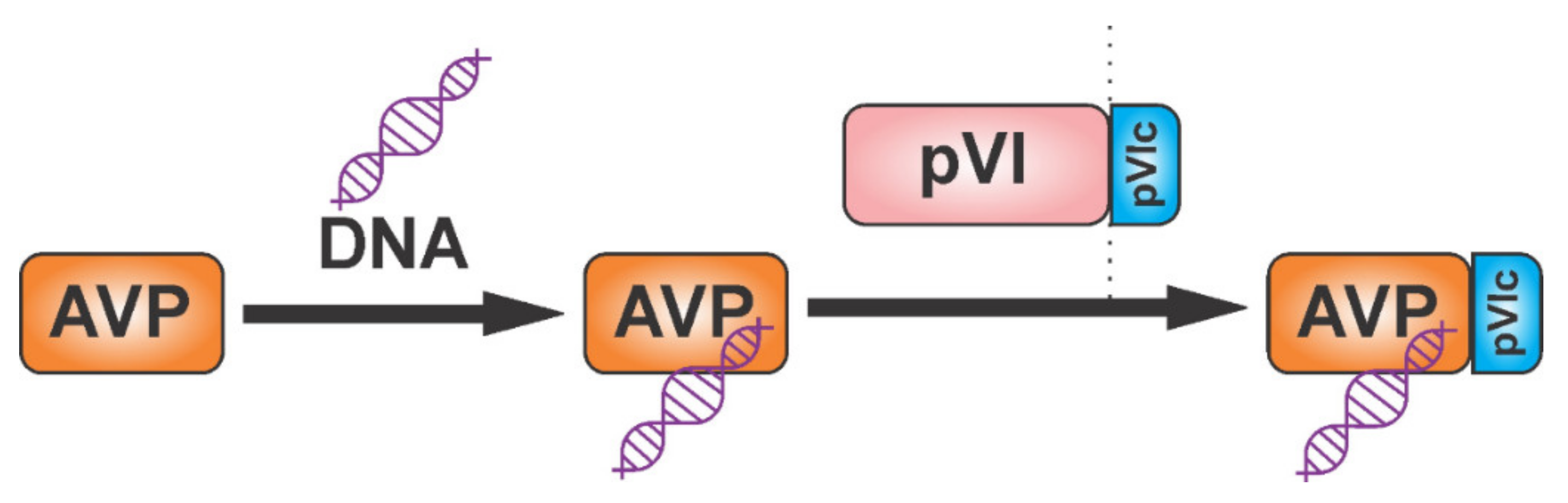
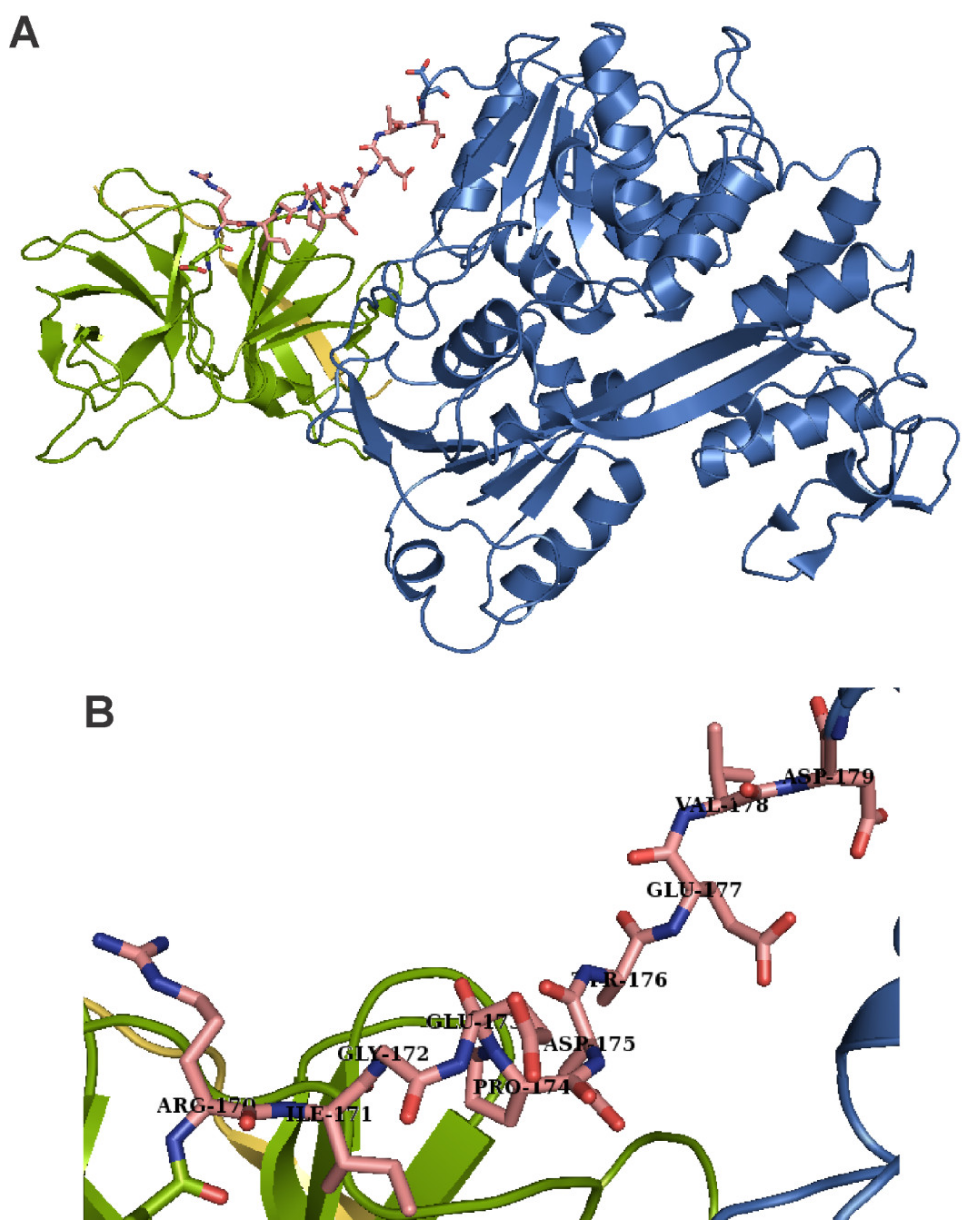
Adenoviruses cause illnesses with symptoms similar to the common cold. They are non-enveloped DNA viruses that combine complex strategies of pre-mRNA splicing and protein processing to produce mature viral proteins [51,52,53,54]. The adenoviral protease adenain is translated from mRNA transcribed from the late L3 gene, together with two proteins important for assembly, namely pre-pVI and hexon. The adenoviral protease is activated by DNA and by the GVQSLKRRRCF peptide, which form a disulfide bond with adenain. This activating peptide is generated by proteolytic cleavage of the pre-pVI protein C-terminus [55,56] (Figure 2). The activation peptide binds far away from the active site. It triggers a bifurcated series of consecutive conformational changes involving 62 amino acids, leading to loop rearrangements and to the long-range communication of both the catalytic histidine and the phenylalanine of the activating peptide [57]. Adenain can cleave eight amino acids from the C-terminus of actin and the resulting peptide acts as an activator in a similar way to the viral activating GVQSLKRRRCF peptide [58].
When a synthetic version of the activating GVQSLKRRRCF peptide was added to adenovirus-infected cells in cell culture, the extent of virus production decreased [59]. This suggests that not only protease inhibition but also premature activation or dysregulation of the protease can be detrimental for the virus.
3. RNA Viruses
3.1. Retroviruses
Retroviruses are enveloped positive single-stranded RNA viruses that use retroviral reverse transcriptase to form a DNA intermediate and then integrase to incorporate the viral DNA into the host genome. Human immunodeficiency viruses (HIV-1 and HIV-2) and human T-lymphotropic viruses (HTLV-1 and HTLV-2) cause lifelong chronical infections in humans [60,61,62]. Fragments of various retroviruses found in the human genome document a wide range of past retroviral infections during history and evolution [63].
The epidemic of HIV in the 1980s led to an intensive effort in the field of antiviral drug development. The first antivirals in clinical use were nucleoside derivatives interfering with the synthesis of proviral DNA, the first being specifically azidothymidine (zidovudine) in 1987 [64,65,66]. The first antivirals with a completely novel mechanism of inhibition were inhibitors of the HIV protease. The earliest of them, saquinavir, was approved by the U.S. Food and Drug Administration (FDA) in 1995 [67,68,69]. The simultaneous targeting of two different steps of viral replication (the synthesis of proviral DNA and the cleavage of viral polyproteins into functional proteins) started a new era in the treatment of AIDS/HIV: the highly active antiretroviral therapy (HAART). Compounds blocking other steps of the viral life cycle followed [70]. Cabotegravir [71,72], a strand transfer HIV integrase inhibitor [73] approved in 2021, is the most recent one among them. HAART changed HIV infection from a fatal to a chronic manageable disease, although the virus remains integrated in host cells. To minimize the risk of evolution of drug-resistant variants [74,75,76,77,78,79,80,81,82], sophisticated drug combinations and the precise dosage are important [83,84,85]. The treatment in individual patients must sometimes be changed not only due to the development of drug resistance but also due to unwanted side effects appearing after long-term therapy [86]. Thus, new compounds with different chemical structures, diverse binding modes, or with novel unique mechanisms of action are continuously desired.
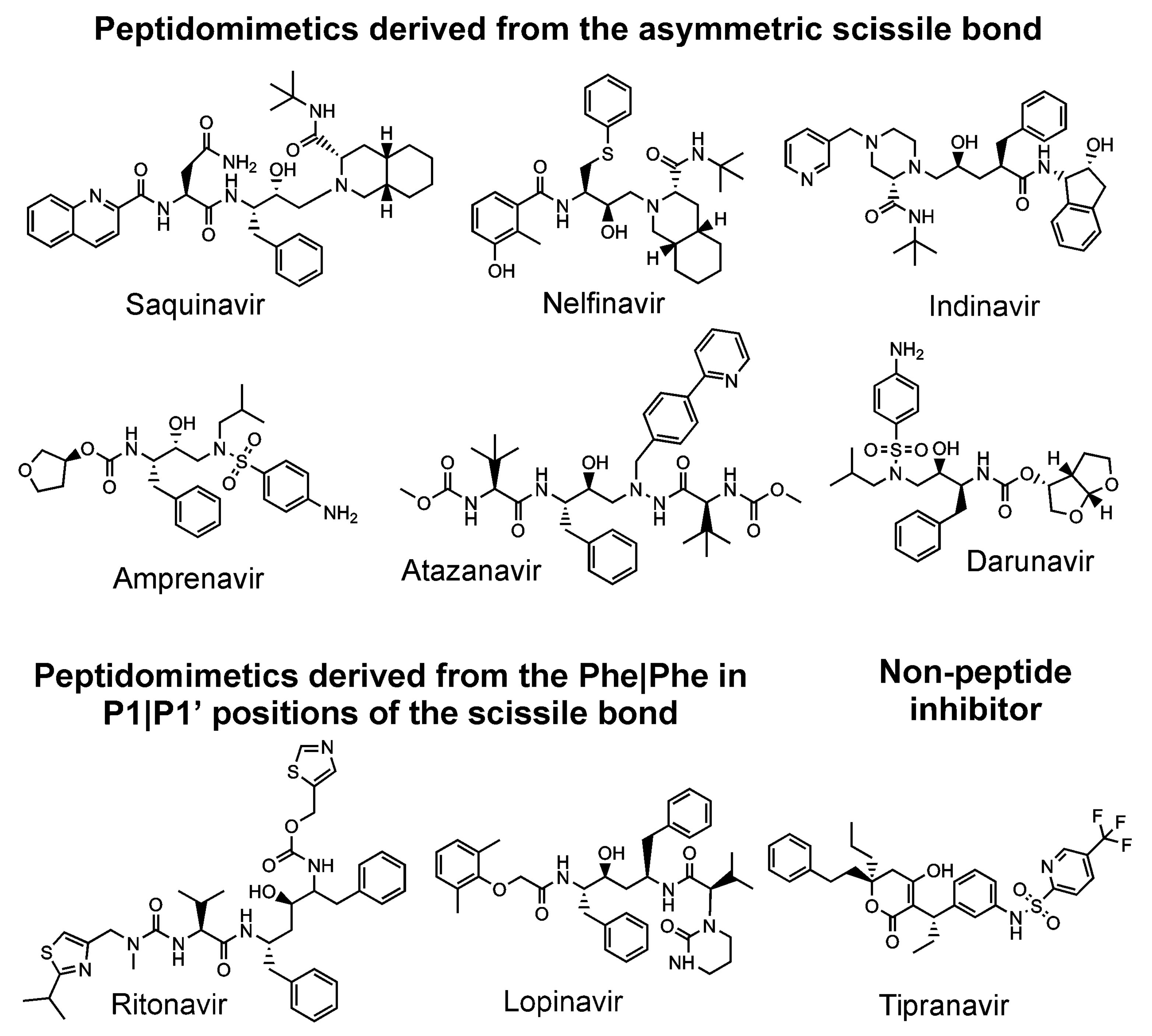
All nine HIV protease inhibitors in clinical use (saquinavir, indinavir, ritonavir, nelfinavir, amprenavir, lopinavir, tipranavir, atazanavir, and darunavir) target the active site [17,79] of the HIV protease, although it has been suggested that darunavir [87] and tipranavir [88] may also block dimerization via a secondary binding site (Figure 3). It has been shown that all these inhibitors have an affinity for the precursor, which is of several orders of magnitude lower than the affinity for its mature form [89,90,91,92]. Thus, an inhibitor targeting the precursor would block the first rate-limiting step of the protease cleavage and maturation cascade [93]. Such a compound could have a higher barrier to drug-resistance development due to its expected ability to conformationally bind loose structures such as precursors and drug-resistant mutants [94]. Darunavir is the most potent inhibitor of the precursor forms of the HIV protease [89,92,95,96]. At the same time, this compound has a very high barrier against drug-resistance development [97].
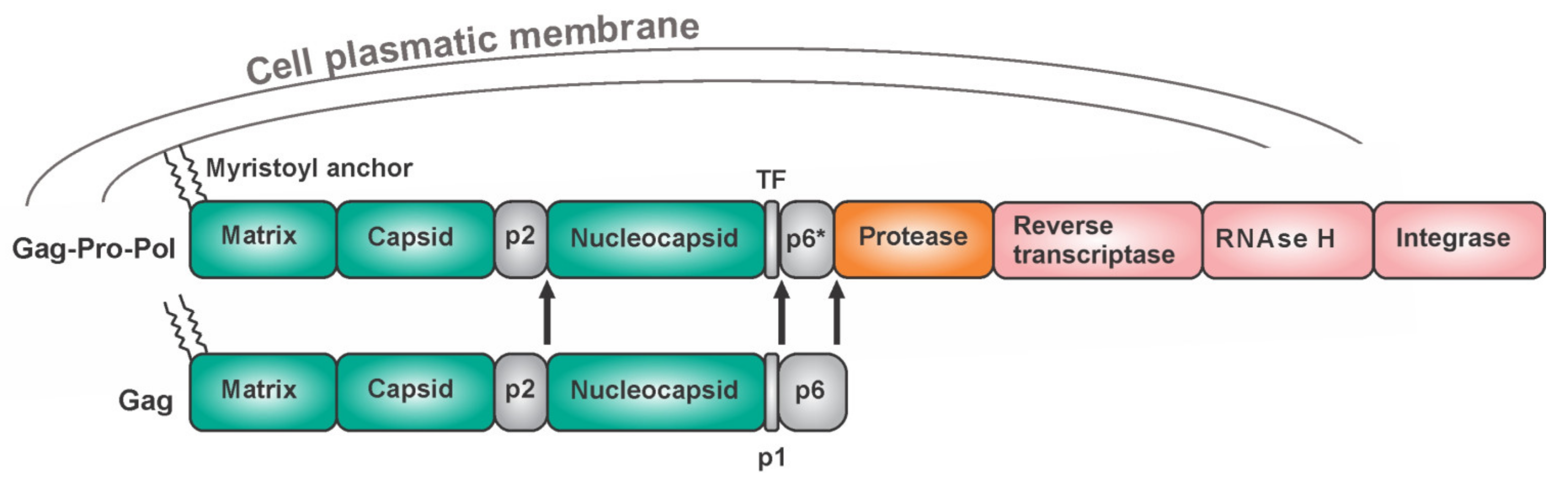
The HIV protease is an obligatory homodimer. Attempts to design dimerization inhibitors have not revealed any clinical candidates [98,99,100,101,102,103]. The HIV protease belongs to the family of aspartic proteases. Each monomer provides one catalytic triad, namely Asp-Thr-Gly, which forms the active site through a network of hydrogen bonds, an arrangement which is referred to as the “fireman grip” [104,105]. The HIV protease is expressed in vivo as part of the Gag-Pol polyprotein (Figure 4). The Gag region harbors structural proteins (matrix, capsid, and nucleocapsid), while the Pol region bears viral enzymes. Gag is expressed separately approximately 95% of the time. After a-1 ribosomal frameshift, Gag is produced in fusion with Pol. This occurs in 5% of the translating events. Keeping the optimal Gag to Gag-Pol ratio is critical for the successful production of viral progeny [106]. One monomer of the HIV protease is embedded in each Gag-Pol macromolecule. To cleave itself out of the precursor, two Gag-Pol moieties must form at least a transient dimeric structure to form the active site. The initial cleavage does not release the mature protease but instead, the first hydrolysis reactions occur intramolecularly (in cis) between the p2 peptide and the nucleocapsid, and between the transframe peptide (TFP) and the p6* peptide, followed by cis N-terminal removal of HIV-1 PR from the precursor (Figure 4). The subsequent steps of protease maturation are intermolecular (trans cleavage) [107,108,109]. The HIV protease occurs in several forms—as the precursor as partially processed polyproteins and as the mature form. In general, partially processed and unprocessed peptides of amino acid sequences found adjacent to the protease region might influence the protease domain, thus affecting its dimerization ability [109,110], substrate specificity, accessibility of cleavage sites, structural features, and stability. Among other retroviruses, as many as three C-terminally truncated mature forms of Mason–Pfizer monkey virus (M-PMV) protease were reported to exist and these have different levels of activity and stability [111,112,113,114,115].
Protease-inhibitor resistant variants harboring mutations not in the protease but in the Gag region [80,116] have been reported and protease mutants change the susceptibility to bevirimat, which is a maturation inhibitor that binds the Gag polyprotein [81]. Gag and Gag-Pol most probably contain intrinsically disordered regions that enable their functional plasticity [117]. The precursor forms may include transient ligand binding sites, which would disappear after maturation and could be targeted by novel drugs.
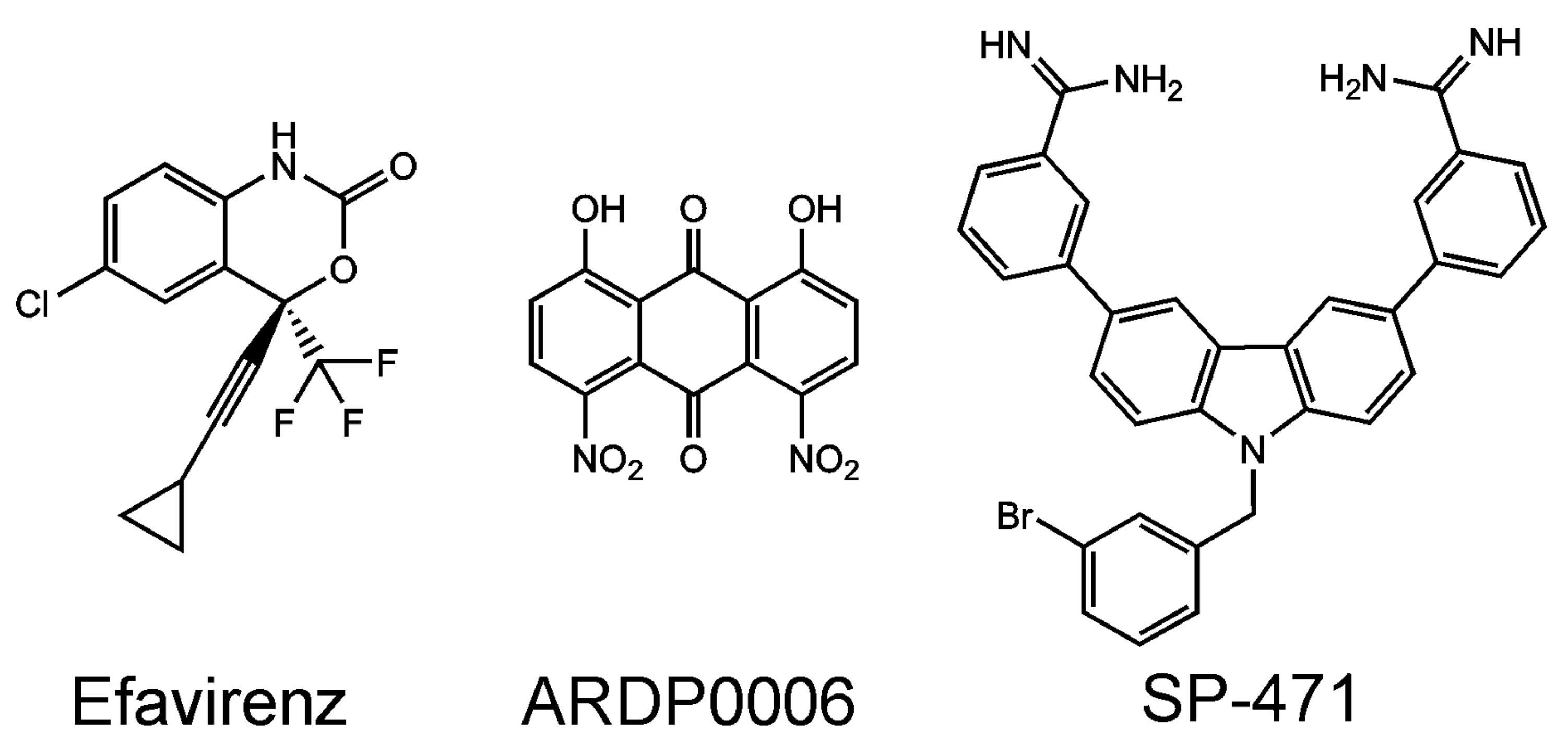
Some non-nucleoside HIV reverse transcriptase inhibitors induce premature HIV protease activation [118]. Namely, efavirenz (Figure 5)—a reverse transcriptase allosteric inhibitor—has an adverse effect on the production of viral particles. It works by bringing the reverse transcriptase domains of two Gag-Pol molecules into close proximity, which is followed by the dimerization of the two protease domains and ultimately leads to premature autoprocessing of HIV protease. Premature cleavage of the Gag-Pol molecules impairs the production of viral particles [119]. This effect was observed in the micromolar concentration range in tissue culture. Such a high concentration of efavirenz is probably well above a safe therapeutic concentration and, moreover, is unlikely to be reachable in vivo. Thus, this finding is not directly applicable in therapy. It represents a valuable proof-of-concept, encouraging the search for more efficient compounds triggering the premature activation of the HIV protease as a completely novel class of drugs. The HIV protease is cytotoxic [92,120,121,122,123] and its premature activation in the cytoplasm of host cells could lead to the elimination of infected cells with HIV integrated in their genome [124,125], thus acting as a causative cure. It is also known that allosteric integrase inhibitors, promoting multimerization of HIV integrase, impair not only integration but also particle core maturation as well as reverse transcription during the subsequent round of virus infection [126]. The influence of allosteric integrase inhibitors on the HIV protease and its processing was not studied to date.
Highly active HIV protease variants seem to be evolutionarily unfavorable. Viral constructs bearing two copies of the HIV PR monomers genetically tethered in one Gag-Pol polyprotein do not produce infectious viral progeny since the premature processing of the viral polyproteins prevents viral particle formation and infectivity. Premature processing also leads to increased cell toxicity [127]. Similarly, the placement of leucine zippers at the C-terminus of HIV PR in viral constructs to force the formation of enzymatically active PR dimers significantly reduced the production of virus-like particles. The production of virus-like particles was restored to wild-type levels by the addition of HIV PR inhibitors [128]. Deletion of the transframe region—a sequence adjacent to the N-terminus of the HIV protease, preventing premature HIV PR autoprocessing—led to the production of virions with improperly processed polyproteins and greatly reduced viral infectivity [129]. Impaired capsid assembly after premature viral protease activation was also reported for Mouse Mammary Tumor Virus [130].
Gag [131,132] and Gag-Pol polyproteins are recruited to the cell membrane, which is the site of viral assembly [133]. In HIV, the N-terminal myristoyl moiety of Gag anchors viral proteins to the cell membrane. Inhibition of myristoylation [134] or blocking of the interaction of viral polyproteins with the host cell membrane could prevent proper virion assembly [135]. It could also mediate in-cell protease activation, supporting the elimination of virus-infected cells through the cytotoxicity of the HIV protease.
3.2. Picornaviruses
Picornaviruses are the causative agents of diseases such as polio, rhinitis, or hepatitis A [136]. They belong to the family of positive-strand RNA non-enveloped viruses and possess a chymotrypsin-like protease with a catalytic cysteine in its active site. This protease is denoted as the 3C proteinase or picornain [137]. It co-translationally and post-translationally cleaves the nascent viral polyprotein. The rate and order of hydrolysis events are precisely tuned and are linked to viral replication. Additionally, not only do the fully processed mature viral proteins have specific functions but the partially cleaved intermediates also play a distinct role, which is indispensable in the viral life cycle.
Picornaviral polyprotein P3 harbors four proteins: 3A [138] (hijacks host factors), 3B (important for priming of RNA synthesis), 3C (protease), and 3D (RNA polymerase). The 3C protease possesses a protease domain with an RNA-binding domain on its surface. The 3CD precursor (fusion of the protease and RNA polymerase) has proteolytic activity, which is several orders of magnitude higher than that of the mature 3C protease when measuring the rate at which structural proteins and 3CD are processed. The other non-structural proteins are cleaved by 3C and 3CD with a comparable efficiency [139]. Although the X-ray structure of mature 3C, 3D, and the precursor 3CD do not show distinct differences between the processed and unprocessed forms [140], changes in the conformational dynamics between free 3C and 3D proteins compared with 3CD have been observed [141]. Synthesis of viral RNA is primed by a covalent link between viral RNA and the 3B protein (also denoted as VPg). P3 molecules recruit other P3 molecules to the replication complex. VPg and the 3D RNA polymerase, which is active only in its active form, are then released by proteolytic cleavage, enabling the synthesis of viral RNA [142]. Interaction of the 3C protease with RNA and with the VPg protein resulted in conformational changes of the RNA-binding site and the active site, indicating long-range allosteric communication between these two sites [143]. Studies with inhibitory antibodies revealed two potential allosteric binding sites, which are conserved and could be used for further drug design [144]. Allosteric compounds and protein–protein/RNA interaction disruptors can represent interesting strategies for picornain-targeting.
The initial cleavage of the full-length viral polyprotein can be catalyzed in cis by a separate protease 2A. This strategy is employed by some picornaviruses of the Rhinovirus and Enterovirus genera. The 2A protease is also a chymotrypsin-like cysteine proteinase and plays a role in the evasion of the host immune response [145]. Viruses belonging to the Aphtovirus and Cardiovirus genera perform the first cleavage using a non-enzymatic mechanism [146].
3.3. Caliciviruses
Caliciviruses cause gastroenteritis in humans [147] and share similar features with picornaviruses. Their viral polyprotein harbors the NF6 protease, which generates partially processed fragments with specific and temporally defined roles in the life cycle, along with mature proteins [148,149,150]. This strategy of using multipurpose proteins is useful for saving genomic space. From the point of view of drug design, targeting one protein can disrupt several different steps in the life cycle.
3.4. Togaviruses
Togaviruses have two genera: Rubivirus (harboring the causative agent of rubella) and Alphavirus. Alphaviruses are positive-enveloped RNA viruses that cause a variety of neglected tropical diseases, including chikungunya [151]. Their viral non-structural proteins are expressed as polyprotein NSp1-NSp2-NSp3 or NSp1-NSp2-NSp3-NSp4 when the stop codon between NSp3 and NSp4 is suppressed [152]. The protease is located in the C-terminus of NSp2 together with an N-terminal helicase. The first cleavage event during protease maturation occurs in cis or in trans between NSp3 (macrodomain) and NSp4 (RNA polymerase), and is followed by the release of NSp1 (mRNA capping enzyme [153]), solely cleaved in cis [154]. In addition to having RNA-modifying activity, NSp1 functions to anchor the replication complex to the host cell membrane and helps the formation of membrane vesicles, which protects the viral replication process from being blocked by the host cell defense machinery. NSp1 monomers associate into a ring structure formed by dodecamers [155]. It is possible that oligomeric structures are also formed by the unprocessed or partially processed precursor polyproteins and could thus drive both the formation of virus-induced membrane microcompartments and the synthesis of viral RNA [155,156]. The complex of NSp1, NSp4, and the fusion protein NSp2-NSp3 synthesizes a negative RNA intermediate [157,158]. The last proteolytic cleavage occurs between NSp2 and NSp3, releasing fully mature enzymes and probably changing the conformation of the RNA-binding surface. This structural change switches the replication complex to synthesize the positive RNA strand. The cleavage between NSp2-NSp3 is driven by a steric change, making the scissile bond accessible. Mutations in NSp3 at the sites of contact with NSp2 lead to decreased production of viral RNA [159]. Targeting of the key protein–protein interactions by small molecule compounds (specifically between NSp2 and NSp3) could disrupt viral replication. Artificial mutations in cleavage sites identified variants that were cleaved more efficiently than wild-type variants [160]. An overactive NSp2 protease variant has been reported [158]. Accelerated processing of viral polyproteins, either due to mutations in the cleavage sites or due to increased enzyme activity, led to a decrease in the production of infectious virions [158,160,161]. This suggests that the optimal cleavage rate does not equate to the maximal cleavage rate and that the natural sequence of events represents a compromise between the cleavage rate and other factors required for successful viral production.
3.5. Flaviviruses
Flaviruses are enveloped positive RNA viruses. Dengue, Zika, West Nile, yellow fever, and tick-borne encephalitis viruses are all members of this family. A more distantly related member of this family is the hepatitis C virus (HCV).
The genetic information of flaviviruses is translated into a single polyprotein harboring three structural (capsid C, membrane precursor prM, and envelope E) and seven non-structural proteins with enzymatic or accessory functions [162]. The viral polyprotein is threaded back and forth through the membrane of the endoplasmic reticulum. The co-translational and post-translational cleavage of the polyprotein is performed by host proteases on the luminal side (at the prM/E, E/NS1, NS1/NS2A, and NS4A/NS4B junctions) [163,164] and by the viral NS2B/NS3 protease on the cytosolic side (at the C/prM, NS2A/NS2B, NS2B/NS3, NS3/NS4A, and NS4B/NS5 junctions) [165,166]. For capsid formation to occur, exact temporally coordinated orchestration of the polyprotein cleavage by the NS2B-NS3 viral protease and by host proteases is necessary [167,168]. Immature virions are formed in the Golgi apparatus, prM is cleaved by furin, and envelope glycoproteins undergo a rearrangement to complete viral maturation [169,170,171].
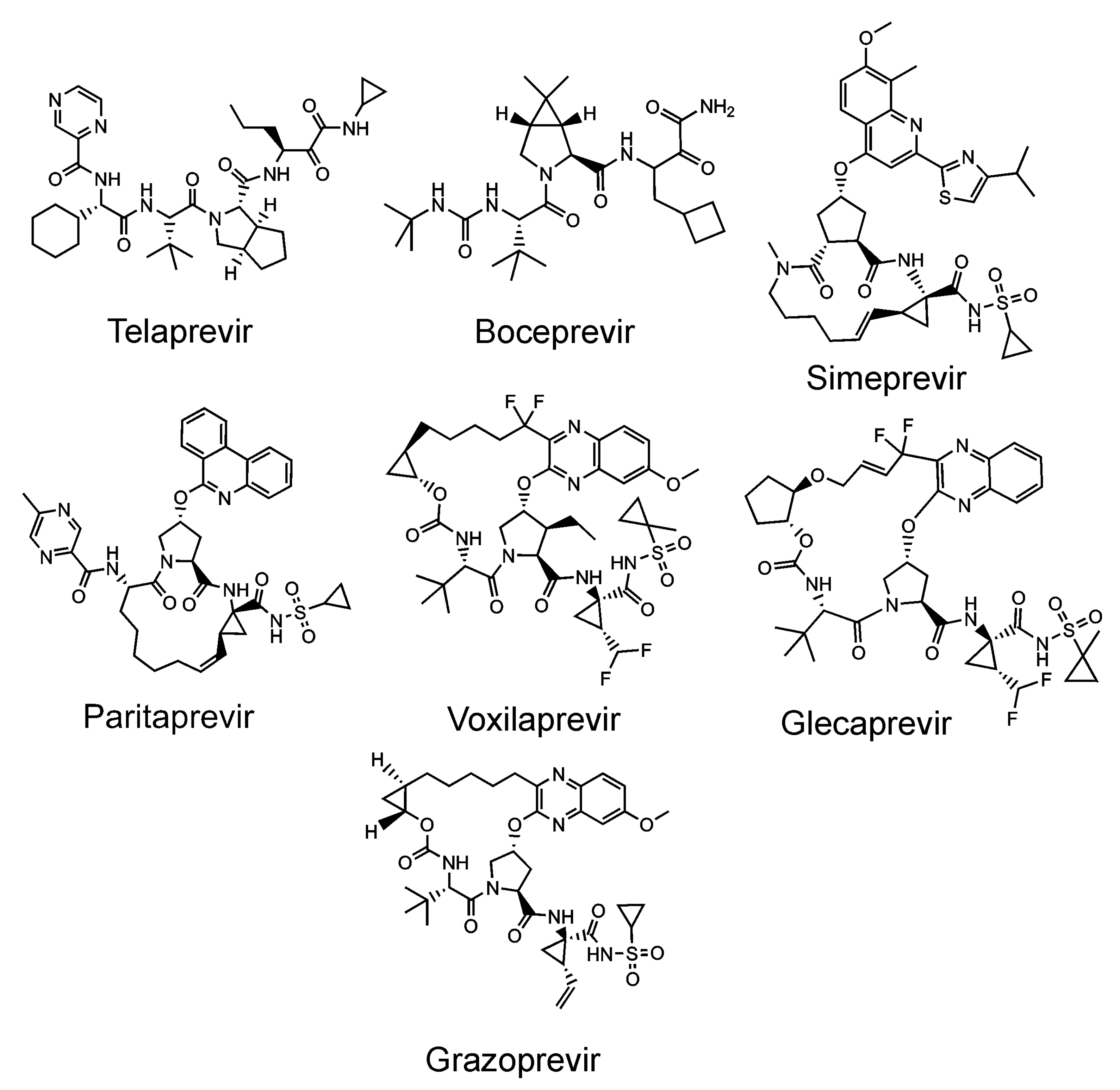
The flaviviral protease consists of the NS3 catalytic domain and the NS2B peptide cofactor for viruses of the Flavivirus genus [165], or the NS4A cofactor in the case of HCV (of the Hepacivirus genus) [172]. The introduction of inhibitors targeting the HCV protease (Figure 6) was a breakthrough in hepatitis C therapy [27,173]. These inhibitors are peptidomimetics targeted against the active site. As with other antivirals, the development of drug resistance represents a potential threat. Inhibitors of no other flaviviral proteases have been introduced into the clinic to date [174,175].
The flaviviral chaperon-like activating peptide NS2B protrudes from the ER membrane, helping to form a chymotrypsin-like structure within the NS3 protease. [176]. A residual activity of solitary NS3 was reported [177]. The catalytic Ser-His-Asp triad is located in the cleft between two β-barrels [178]. Cleavage between the NS2B and NS3 proteins may not be necessary for proper activity, as during in vitro studies, the NS2B-NS3 protease was active as a fusion protein connected with a polyglycine linker between the NS2B and NS3 domains [179,180]. Compared to the NS2B-NS3 fusion protease, the non-covalently associated heterodimer formed by the NS2B and NS3 subunits might be oriented differently towards the cell membrane or may have some subtle differences in its substrate preference, which may be important for orchestrated polyprotein precursor cleavage [181]. The NS3 protein of flaviviruses contains both the protease and the helicase domains. The helicase domain has NTPase and nucleic acid unwinding activity. Despite the helicase domain having no effect on protease activity, the presence of the protease domain improves RNA unwinding activity in Kunjin virus NS3 [182] as well as in Dengue virus NS3 [183]. ATP hydrolase and RNA triphosphatase activity (catalyzed also by the helicase domain) were influenced by neither the presence nor the absence of the protease domain in Kunjin virus NS3 [182]. The ATPase active site of Dengue virus has lower affinity for ATP when the protease domain is present [184]. This can be explained using the structure of the full-length flaviviral NS3 (Figure 7). The connection between the protease and helicase domains is in contact with the NTPase domain active site, which is blocked by the linker between the protease and helicase. This might result in the reduced accessibility of the active site for ATP [180].
Since the active site of the flaviviral protease is flat and would need to be inhibited with charged ligands, whose transportation across the cell membrane is likely to be problematic, alternative antiviral strategies could be used, such as allosteric inhibitors [185] or disruptors of protein–protein interactions.
Apart from the canonical interprotein cleavage sites, another sequence of the intramolecular cleavage event is needed for the temporal orchestration of the viral life cycle [186,187]. It has been shown that at least three cleavages occur exclusively in cis (i.e., intramolecularly) at sites flanking the NS3 and at an internal cleavage site within the NS3 helicase. When the intrahelicase cleavage site was blocked by mutagenesis, the accumulation of unprocessed precursors inhibited the production of infectious viral particles in trans (i.e., intermolecularly). The inhibitory effect was trans-dominant when cells were co-infected by wild-type mutants and by an autoprocessing defective mutant [188]. ARDP0006 (1,8-dihydroxy-4,5-dinitroanthraquinone), an inhibitor of the Dengue virus-2 (DENV-2) protease, has been reported to inhibit virus replication in tissue culture in the low micromolar range [189]. Using in vitro enzyme kinetic measurements with purified enzymes, ARDP0006 was several orders of magnitude weaker than in the tissue culture [190]. Experiments have shown that ARDP0006 has a much higher affinity for the precursor form of the DENV-2 than for its mature form and that this compound preferentially blocks intrahelicase cis-cleavage. The accumulation of unprocessed precursors inhibited the production of infectious viral particles in trans (intermolecularly). The accumulation of precursors also blocked the production of mature virions in trans. Thus, the inhibitory effect of ARDP0006 can persist even if a cell harbors a mix of susceptible and ARDP0006-resistant viral variants [188].
Multimodal inhibitors with the carbazole scaffold, which block cis as well as trans cleavage events by the Dengue protease, have been reported [191] (Figure 5). Targeting of the protease precursor, exclusively autoprocessed in cis, represents an interesting strategy for the counter-development of drug resistance.
Another approach used the Dengue protease as a suicide initiator to activate a prodrug, resulting in the release of the cytotoxic combretastatin. This led to selective killing of virus-infected host cells [192]. Suicide protease substrates represent another alternative therapeutic strategy useful for the elimination of virus-infected cells.
3.6. Coronaviruses
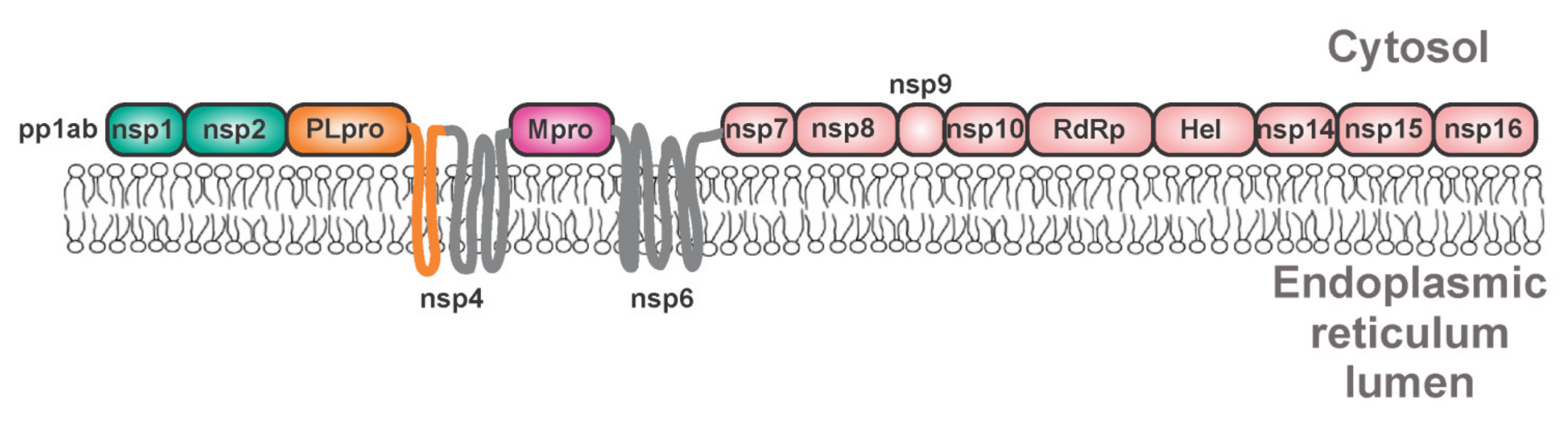
Coronaviruses are detrimental human and animal pathogens [194]. These single-stranded positive RNA-enveloped viruses cause respiratory, enteric, neurological, and hepatic diseases. Including novel SARS-CoV-2, seven human pathogenic coronaviruses were identified [195]. Coronaviruses possess four major structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N), all translated from subgenomic RNAs [196]. The viral RNA is about 29.8 kb long and is translated into two polyproteins, namely pp1a and pp1ab [197] (Figure 8). These polyproteins are cleaved by two or three viral cysteine proteases, by the chymotrypsin-like main protease (Mpro, 3CLpro, and nsp5), and by one or two papain-like proteases. SARS-CoV-2 possesses one of each type of these proteases. The papain-like protease (PLpro and nsp3) releases the first three proteins: nsp1, nsp2, and nsp3 [198]. Mpro releases the remaining 13 non-structural proteins (including itself) [199]. The viral non-structural proteins are produced as polyprotein 1a (bearing proteins nsp1–nsp5) or polyprotein 1ab, which result from a frameshift (Figure 8) [200].
Mpro is activated via dimerization and its monomeric form is nearly inactive [201]. Oligomerization can further increase activity as seen during in vitro studies with a highly active octamer of Mpro [202]. Despite its dimerization, molecular dynamics studies suggest that only one protomer is active within the Mpro dimer [203]. Both active sites of the dimer are occupied by inhibitors in X-ray structures. Symmetrical as well as asymmetrical 3D structures of dimers were reported for the SARS-CoV-2 Mpro complex with an inhibitor. In the asymmetric dimer, one monomer exhibited inactive conformation of the active site [204,205]. The assumption of asymmetry of the SARS-CoV-2 Mpro dimer was supported by molecular dynamics simulations [206] and implies allosteric cooperativity between subunits [205,207].
The release of the mature protease from the precursor is initiated at the N-terminus of Mpro, presumably in cis in an intra-dimer and inter-protomer manner (i.e., two unprocessed monomers form a dimer and one monomer cleaves the second monomer), or in trans, later followed by C-terminal cleavage occurring in trans [208,209,210,211,212,213]. Structural “snap-shots” of cleavage and dimerization events were obtained during crystallization trials [211,213]. Amino acids adjacent to the N-terminus of Mpro influence the conformation of the active site by preventing dimerization, thus diminishing its activity. Potentially druggable ligand binding sites were identified in the precursor, which do not occur in the mature enzyme [211]. Studies with artificial peptide substrates based on SARS-CoV natural cleavage sites showed that the peptides corresponding to the cleavage sites adjacent to Mpro are hydrolyzed much more efficiently than peptides derived from the remaining cleavage sites [214]. The same conclusion was reached when calculating catalytic efficiency from data obtained from in vitro assays with N and C-terminal mutants [209]. These mutants, designed as uncleavable pro-forms of the wild-type enzyme with mutated N and/or C-autoprocessing sites, cleaved a proteolytically inactive precursor with authentic N and C-terminal flanking regions. The mature protease was inhibited by the precursor form with an active site mutation due to cross-dimerization of the mature and mutated pro-form of the protease [209].
For SARS-CoV, it has been found that dimerization-defective mutants (Arg4Glu, Glu290Arg, and Arg298Glu) can be autoprocessed on the N-terminus in cis but are not active in trans in vitro. An active-site mutant of the dimerization-defective variant was cleaved by the mature dimerization-defective mutant, indicating that N-terminal autoprocessing requires only a transient dimerization without a fixed conformation of the mature active site [210]. In particular, mutation of Arg298 results in a monomeric form of Mpro, leading to an inactive conformation and eventually to irreversible collapse of the substrate pocket [215]. The key residue connecting the substrate binding and dimerization events is Glu166 [216]. This residue is responsible for recognizing Gln in the P1 position of substrates and for the interaction with the S1 pocket of the heterologous protomer [208]. Dimerization-defective mutants can presumably be stabilized after substrate binding. When Glu166 is mutated, this mode of dimerization vanishes [216]. Dimer stabilization by substrates was reported also for other viral proteases [217,218].
Hyperactive variants of SARS Mpro have been reported. The mutations of Ser284-Thr285-Ile286 and Phe291 to Ala lead to increased enzyme activity without an apparent effect on its structure and dimerization. It was concluded that these critical residues form a nano-channel acting as an allosteric regulator [219,220,221].
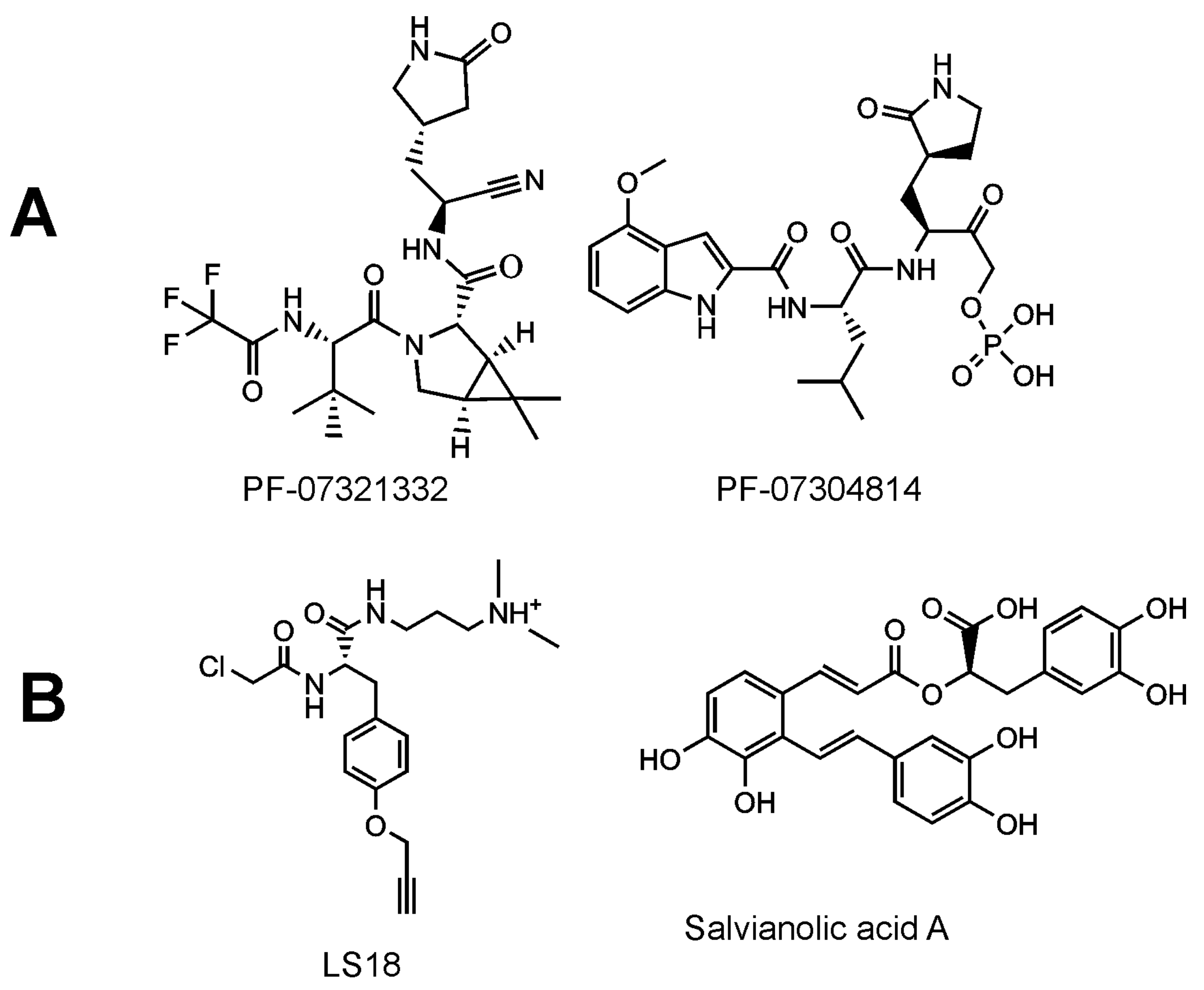
Inhibitors of SARS-CoV-2 targeting the Mpro improved outcomes in a mouse model of coronaviral infection and increased survival of mice [222]. Potent inhibitors of SARS-CoV-2 have been reported, including GC376, a bisulphite adduct of a peptide aldehyde (preclinical studies have been initiated by the Anivive Lifesciences Company, Long Beach, CA, USA) [223,224], and a hydroxymethylketone derivative, specifically PF-07304814, which is an oral drug candidate of Pfizer [225] (Figure 9). The clinical candidates are prodrugs. Upon activation, the inhibitors are targeted against the Mpro active site, forming a covalent bond between the catalytic nucleophilic cysteinevia an electrophilic warhead. The key structural feature seems to be the γ-lactam glutamine surrogate at the P1 position. Recently, another inhibitor of Mpro, denoted as PBI-0451, has entered into phase 1 clinical trials (ClinicalTrials.gov registration number: NCT05011812).
Various inhibitors and strategies are under investigation. The attention was first focused on the potential repurposing of approved medicines [226,227,228] and masitinib, a tyrosine kinase inhibitor, has been identified among them [229]. Another strategy is represented by de novo designs of active-site [230,231] and allosteric ligands [232]. Natural products, such as flavonoids and their derivatives [233,234] or terpenes, were shown to be inhibitors of Mpro. For example, in addition to its inhibitory activity at submicromolar concentrations, the terpene eugenol promoted the oligomerization of Mpro in vitro [235].
The evaluation of the effect of Mpro inhibitors on the precursor forms of the protease has not yet received sufficient attention. Inhibition of the pre-processed Mpro has been attempted in vitro using a non-cleavable N-terminal Strep-tagged variant of Mpro as a model precursor. Click chemistry was used to fluorescently label the ligands covalently bound to the active site. Next followed SDS (sodium dodecyl sulfate) electrophoresis to separate the inhibitor-bound model precursor of Mpro and the fluorescence intensity of the band corresponding to the model precursor of Mpro was evaluated. A compound derived from tyrosine, harboring a chloroacetamide warhead, was identified as a precursor inhibitor. This compound was used as a probe in a competitive screen and salvianolic acid A was evaluated as a most-potent compound inhibiting in the low-micromolar range (Figure 9) [236]. The active site of SARS-CoV-2 Mpro seems to be structurally malleable [236,237,238], a feature which could be more pronounced in the case of the precursor forms. Targeting the unprocessed protease would block the maturation process during the first irreversible step. Even if such an inhibitor was removed, this might not lead to a recovery of viral maturation, as indicated by experiments with the HIV protease [239]. Dimerization inhibitors and allosteric ligands binding to the precursor form could also be interesting first-in-class drugs targeting SARS-CoV-2 Mpro.
In beta coronaviruses, a large 213 kDa nsp3 harbors a 36 kDa papain-like protease. This protease self-releases nsp3 out of the viral polyprotein and cleaves host protein substrates [240]. For self-cleavage at the nsp3/4 boundary, membrane association of nsp3 might be required [241]. The proteases probably self-cleave later in the coronaviral life cycle because the intermediates nsp2-nsp3 and nsp4-nsp11 were identified in SARS-CoV and Mouse hepatitis virus-infected cells [241,242]. Intermediates between the precursor and the mature protein are also predicted to be participants in the viral replication cycle [241,243].
The coronaviral papain-like protease (PLpro) possesses specific structural features, namely the palm, thumb, finger (containing a zinc ion), and a ubiquitin-like domain [244]. The finger and palm domains are important for interactions with ubiquitin, with this interaction being the first step of deubiquitylation activity [245]. The papain-like protease also releases the ISG15 protein, thus modulating the innate host immune response [246]. PLpro can also stabilize an E3 ubiquitin ligase and help ubiquitinate p53 [247]. Predicted inhibitors of PLpro, such as disulfiram [248,249] (ClinicalTrials.gov registration number: NCT04485130) and isotretinoin (ClinicalTrials.gov registration number: NCT04361422), are both in Phase 2 clinical trials due to their potential for repurposing.
A possible overlap of coronaviral papain-like proteases with substrate specificities of host deubiquitylating enzymes makes it difficult to design active-site inhibitors. Highly specific active-site inhibitors are therefore desired [250]. Allosteric modulators and other compounds with alternative mechanisms of action could be more suitable to avoid off-target binding. Yeast-surface display nanobodies (a single-domain antibody) were developed as potential inhibitors of PLpro [251]. This strategy represents an interesting alternative to classical inhibitors.
4. Concluding Remarks
Inhibitors of HIV and HCV proteases are important components of current antiviral therapies. These drugs primarily bind to the enzymes’ active sites. Proteases from other viruses can be exploited for therapeutic intervention as well.
Targeting viral proteases in their precursor form brings several possibilities for the development of compounds with unique mechanisms of action. Inhibitors binding to the precursor form would block the first rate-limiting step of the “domino” cascade, leading to viral maturation. Such compounds would bind to the active site of a precursor (which can differ in terms of the substrate preferences and/or in the structural stability to that of the mature form) or to other binding sites of interacting partners or natural modulators. Transiently occurring allosteric binding sites of the precursor represent another interesting alternative. It is not only the inhibition but also the up-regulation or premature protease activation that is detrimental for the virus. Viral proteases are often cytotoxic and thus their increased activity in infected cells could help to eliminate virus-bearing host cells. Such a possibility is interesting mainly for chronically infecting viruses. Regardless of the chosen approach, viral protease precursors offer a promising frontier for drug discovery.
Author Contributions
Conceptualization, T.M.; resources, T.M., P.N.; writing—original draft preparation, T.M, P.N.; writing—review and editing, T.M., P.N.; figures preparation, P.N.; supervision, T.M. All authors have read and agreed to the published version of the manuscript.
Funding
Project Chemical Biology for Development of Novel Therapies, European Regional Development Fund; OP RDE, CZ.02.1.01/0.0/0.0/16_019/0000729.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We want to thank Jan Konvalinka for his support, Blanka Collis for the extremely helpful text editing, and Joshua Smith for the language corrections and inspiring comments. This work was supported by European Regional Development Fund; OP RDE, CZ.02.1.01/0.0/0.0/16_019/0000729, Project Chemical Biology for Development of Novel Therapies.
Conflicts of Interest
All authors declare no conflicts of interests.
References
- Kassell, B.; Kay, J. Zymogens of Proteolytic Enzymes. Science 1973, 180, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Keil, B.; Meloun, B.; Vanecek, J.; Kostka, V.; Prusik, Z.; Sorm, F. Partial structure of chymotrypsinogen. Biochim. Biophys. Acta 1962, 56, 595–599. [Google Scholar] [CrossRef]
- Meloun, B.; Kluh, I.; Kostka, V.; Morávek, L.; Prusík, Z.; Vanĕcek, J.; Keil, B.; Sorm, F. Covalent structure of bovine chymotrypsinogen A. Biochim. Biophys. Acta 1966, 130, 543–546. [Google Scholar] [CrossRef]
- Keilova, H.; Kostka, V.; Kay, J. The first step in the activation of chicken pepsinogen is similar to that of prochymosin. Biochem. J. 1977, 167, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichová, I.; Kostka, V. Molecular characteristics of pepsinogen and pepsin from duck glandular stomach. Comp. Biochem. Physiol. B 1990, 97, 89–94. [Google Scholar] [CrossRef]
- Strop, P.; Cechová, D.; Tomásek, V. Model study of hydrophobic interactions of alpha- and beta-trypsin and alpha-chymotrypsin. J. Chromatogr. 1983, 259, 255–268. [Google Scholar] [CrossRef]
- Hynek, R.; Kasicka, V.; Kucerová, Z.; Kás, J. Fast detection of phosphorylation of human pepsinogen A, human pepsinogen C and swine pepsinogen using a combination of reversed-phase high-performance liquid chromatography and capillary zone electrophoresis for peptide mapping. J. Chromatogr. B Biomed. Sci. Appl. 1997, 688, 213–220. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Calvo-Rojas, G.; Lecumberri, R.; Rocha, E.; Pozo-Hernández, C.; Terleira-Fernández, A.I.; Vargas-Castrillón, E. Discovery of anticoagulant drugs: A historical perspective. Curr. Drug Discov. Technol. 2012, 9, 83–104. [Google Scholar] [CrossRef]
- Foltmann, B. A review on prorennin and rennin. C. R. Trav. Lab. Carlsberg 1966, 35, 143–231. [Google Scholar]
- Hasilik, A.; von Figura, K.; Conzelmann, E.; Nehrkorn, H.; Sandhoff, K. Lysosomal enzyme precursors in human fibroblasts. Activation of cathepsin D precursor in vitro and activity of beta-hexosaminidase A precursor towards ganglioside GM2. Eur. J. Biochem. 1982, 125, 317–321. [Google Scholar] [CrossRef]
- Mása, M.; Maresová, L.; Vondrásek, J.; Horn, M.; Jezek, J.; Mares, M. Cathepsin D propeptide: Mechanism and regulation of its interaction with the catalytic core. Biochemistry 2006, 45, 15474–15482. [Google Scholar] [CrossRef]
- Hánová, I.; Brynda, J.; Houštecká, R.; Alam, N.; Sojka, D.; Kopáček, P.; Marešová, L.; Vondrášek, J.; Horn, M.; Schueler-Furman, O.; et al. Novel Structural Mechanism of Allosteric Regulation of Aspartic Peptidases via an Evolutionarily Conserved Exosite. Cell Chem. Biol. 2018, 25, 318–329.e314. [Google Scholar] [CrossRef] [Green Version]
- Houštecká, R.; Hadzima, M.; Fanfrlík, J.; Brynda, J.; Pallová, L.; Hánová, I.; Mertlíková-Kaiserová, H.; Lepšík, M.; Horn, M.; Smrčina, M.; et al. Biomimetic Macrocyclic Inhibitors of Human Cathepsin D: Structure-Activity Relationship and Binding Mode Analysis. J. Med. Chem. 2020, 63, 1576–1596. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef]
- Shinde, U.; Fu, X.; Inouye, M. A Pathway for Conformational Diversity in Proteins Mediated by Intramolecular Chaperones. J. Biol. Chem. 1999, 274, 15615–15621. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.S.; Banerjee, S.; Aponte-Diaz, D.; Sharma, S.D.; Aligo, J.; Lodeiro, M.F.; Ning, G.; Sharma, R.; Arnold, J.J.; Cameron, C.E. Multiple poliovirus-induced organelles suggested by comparison of spatiotemporal dynamics of membranous structures and phosphoinositides. PLoS Pathg. 2018, 14, e1007036. [Google Scholar] [CrossRef] [Green Version]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.; Dejnirattisai, W.; Carrique, L.; Martin, I.S.; Karia, D.; Ilca, S.L.; Ho, S.F.; Kotecha, A.; Keown, J.R.; Mongkolsapaya, J.; et al. Flavivirus maturation leads to the formation of an occupied lipid pocket in the surface glycoproteins. Nat. Commun. 2021, 12, 1238. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.; Reggiori, F.; de Haan, C.A. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458–459, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Aleshin, A.E.; Drag, M.; Gombosuren, N.; Wei, G.; Mikolajczyk, J.; Satterthwait, A.C.; Strongin, A.Y.; Liddington, R.C.; Salvesen, G.S. Activity, Specificity, and Probe Design for the Smallpox Virus Protease K7L. J. Biol. Chem. 2012, 287, 39470–39479. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Culp, J.S.; DiLella, A.G.; Hellmig, B.; Hoog, S.S.; Janson, C.A.; Smith, W.W.; Abdel-Meguid, S.S. Unique fold and active site in cytomegalovirus protease. Nature 1996, 383, 275–279. [Google Scholar] [CrossRef]
- Saribas, A.S.; Coric, P.; Bouaziz, S.; Safak, M. Expression of novel proteins by polyomaviruses and recent advances in the structural and functional features of agnoprotein of JC virus, BK virus, and simian virus 40. J. Cell Physiol. 2019, 234, 8295–8315. [Google Scholar] [CrossRef]
- Hejtmánková, A.; Roubalová, K.; Forejtová, A.; Žáčková Suchanová, J.; Forstová, J.; Viklický, O.; Španielová, H. Prevalence of antibodies against BKPyV subtype I and IV in kidney transplant recipients and in the general Czech population. J. Med. Virol. 2019, 91, 856–864. [Google Scholar] [CrossRef]
- Majerová, T.; Hoffman, H.; Majer, F. Therapeutic targets for influenza-Perspectives in drug development. Collect. Czechoslov. Chem. Commun. 2010, 75, 81–103. [Google Scholar] [CrossRef]
- Yip, W.K.W.; Cristi, F.; Trifonov, G.; Narayan, N.; Kubanski, M.; Shmulevitz, M. The reovirus μ2 C-terminal loop inversely regulates NTPase and transcription functions versus binding to factory-forming μNS and promotes replication in tumorigenic cells. J. Virol. 2021, 95, e02006-20. [Google Scholar] [CrossRef]
- Zając, M.; Muszalska, I.; Sobczak, A.; Dadej, A.; Tomczak, S.; Jelińska, A. Hepatitis C—New drugs and treatment prospects. Eur. J. Med. Chem. 2019, 165, 225–249. [Google Scholar] [CrossRef]
- Gable, J.E.; Acker, T.M.; Craik, C.S. Current and Potential Treatments for Ubiquitous but Neglected Herpesvirus Infections. Chem. Rev. 2014, 114, 11382–11412. [Google Scholar] [CrossRef] [Green Version]
- Clercq, E.D.; Sakuma, T.; Baba, M.; Pauwels, R.; Balzarini, J.; Rosenberg, I.; Holý, A. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antivir. Res. 1987, 8, 261–272. [Google Scholar] [CrossRef]
- Snoeck, R.; Sakuma, T.; De Clercq, E.; Rosenberg, I.; Holy, A. (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 1988, 32, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Tsuge, H.; Almassy, R.J.; Gribskov, C.L.; Katoh, S.; Vanderpool, D.L.; Margosiak, S.A.; Pinko, C.; Matthews, D.A.; Kan, C.-C. Structure of the Human Cytomegalovirus Protease Catalytic Domain Reveals a Novel Serine Protease Fold and Catalytic Triad. Cell 1996, 86, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Darke, P.L.; Cole, J.L.; Waxman, L.; Hall, D.L.; Sardana, M.K.; Kuo, L.C. Active human cytomegalovirus protease is a dimer. J. Biol. Chem. 1996, 271, 7445–7449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.; Qian, C.; Massariol, M.J.; Bonneau, P.R.; Cordingley, M.G.; Lagacé, L. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature 1996, 383, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Shieh, H.S.; Kurumbail, R.G.; Stevens, A.M.; Stegeman, R.A.; Sturman, E.J.; Pak, J.Y.; Wittwer, A.J.; Palmier, M.O.; Wiegand, R.C.; Holwerda, B.C.; et al. Three-dimensional structure of human cytomegalovirus protease. Nature 1996, 383, 279–282. [Google Scholar] [CrossRef]
- Liu, F.Y.; Roizman, B. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J. Virol. 1991, 65, 5149–5156. [Google Scholar] [CrossRef] [Green Version]
- Oien, N.L.; Thomsen, D.R.; Wathen, M.W.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Assembly of herpes simplex virus capsids using the human cytomegalovirus scaffold protein: Critical role of the C terminus. J. Virol. 1997, 71, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Sheaffer, A.K.; Newcomb, W.W.; Brown, J.C.; Gao, M.; Weller, S.K.; Tenney, D.J. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J. Virol. 2000, 74, 6838–6848. [Google Scholar] [CrossRef] [Green Version]
- Baum, E.Z.; Bebernitz, G.A.; Hulmes, J.D.; Muzithras, V.P.; Jones, T.R.; Gluzman, Y. Expression and analysis of the human cytomegalovirus UL80-encoded protease: Identification of autoproteolytic sites. J. Virol. 1993, 67, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Holwerda, B.C.; Wittwer, A.J.; Duffin, K.L.; Smith, C.; Toth, M.V.; Carr, L.S.; Wiegand, R.C.; Bryant, M.L. Activity of two-chain recombinant human cytomegalovirus protease. J. Biol. Chem. 1994, 269, 25911–25915. [Google Scholar] [CrossRef]
- Loveland, A.N.; Chan, C.K.; Brignole, E.J.; Gibson, W. Cleavage of human cytomegalovirus protease pUL80a at internal and cryptic sites is not essential but enhances infectivity. J. Virol. 2005, 79, 12961–12968. [Google Scholar] [CrossRef] [Green Version]
- Shimba, N.; Nomura, A.M.; Marnett, A.B.; Craik, C.S. Herpesvirus protease inhibition by dimer disruption. J. Virol. 2004, 78, 6657–6665. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.M.; Shahian, T.; Baharuddin, A.; Gable, J.E.; Craik, C.S. Enzyme Inhibition by Allosteric Capture of an Inactive Conformation. J. Mol. Biol. 2011, 411, 999–1016. [Google Scholar] [CrossRef] [Green Version]
- Shahian, T.; Lee, G.M.; Lazic, A.; Arnold, L.A.; Velusamy, P.; Roels, C.M.; Guy, R.K.; Craik, C.S. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat. Chem. Biol. 2009, 5, 640–646. [Google Scholar] [CrossRef]
- Yamanaka, G.; DiIanni, C.L.; O’Boyle, D.R., II; Stevens, J.; Weinheimer, S.P.; Deckman, I.C.; Matusick-Kumar, L.; Colonno, R.J. Stimulation of the Herpes Simplex Virus Type I Protease by Antichaeotrophic Salts. J. Biol. Chem. 1995, 270, 30168–30172. [Google Scholar] [CrossRef] [Green Version]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef]
- Wang, J.; Loveland, A.N.; Kattenhorn, L.M.; Ploegh, H.L.; Gibson, W. High-molecular-weight protein (pUL48) of human cytomegalovirus is a competent deubiquitinating protease: Mutant viruses altered in its active-site cysteine or histidine are viable. J. Virol. 2006, 80, 6003–6012. [Google Scholar] [CrossRef] [Green Version]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.T.; Oh, S.E.; Lee, Y.O.; Gibson, W.; Ahn, J.H. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 2009, 83, 12046–12056. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-κB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [Green Version]
- Westergren Jakobsson, A.; Segerman, B.; Wallerman, O.; Bergström Lind, S.; Zhao, H.; Rubin, C.-J.; Pettersson, U.; Akusjärvi, G. The Human Adenovirus 2 Transcriptome: An Amazing Complexity of Alternatively Spliced mRNAs. J. Virol. 2021, 95, e01869-20. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.P.; Mathews, M.B. DNA replication and the early to late transition in adenovirus infection. Cell 1980, 22, 523–533. [Google Scholar] [CrossRef]
- Donovan-Banfield, I.a.; Turnell, A.S.; Hiscox, J.A.; Leppard, K.N.; Matthews, D.A. Deep splicing plasticity of the human adenovirus type 5 transcriptome drives virus evolution. Commun. Biol. 2020, 3, 124. [Google Scholar] [CrossRef]
- Pied, N.; Wodrich, H. Imaging the adenovirus infection cycle. FEBS Lett. 2019, 593, 3419–3448. [Google Scholar] [CrossRef] [Green Version]
- Webster, A.; Hay, R.T.; Kemp, G. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell 1993, 72, 97–104. [Google Scholar] [CrossRef]
- Mangel, W.F.; McGrath, W.J.; Toledo, D.L.; Anderson, C.W. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 1993, 361, 274–275. [Google Scholar] [CrossRef]
- Baniecki, M.L.; McGrath, W.J.; Mangel, W.F. Regulation of a Viral Proteinase by a Peptide and DNA in One-dimensional Space: III. Atomic resolution structure of the nascent form of the adenoirus proteinase. J. Biol. Chem. 2013, 288, 2081–2091. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.T.; McBride, K.M.; Baniecki, M.L.; Reich, N.C.; Marriott, G.; Mangel, W.F. Actin can act as a cofactor for a viral proteinase in the cleavage of the cytoskeleton. J. Biol. Chem. 2002, 277, 46298–46303. [Google Scholar] [CrossRef] [Green Version]
- Ruzindana-Umunyana, A.; Sircar, S.; Weber, J.M. The Effect of Mutant Peptide Cofactors on Adenovirus Protease Activity and Virus Infection. Virology 2000, 270, 173–179. [Google Scholar] [CrossRef]
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [Green Version]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Weiss, R.A.; Griffiths, D.J. Human RNA Viruses: The Search for Novel Human Retroviruses in Chronic Disease. Microbiol Mol. Biol. Rev. 2008, 72, 157–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clercq, E. HIV resistance to reverse transcriptase inhibitors. Biochem. Pharmacol. 1994, 47, 155–169. [Google Scholar] [CrossRef]
- Holý, A.; Rosenberg, I. Synthesis of 9-(2-phosphonylmethoxyethyl)adenine and related compounds. Collect. Czechoslov. Chem. Commun. 1987, 52, 2801–2809. [Google Scholar] [CrossRef]
- Balzarini, J.; Holy, A.; Jindrich, J.; Naesens, L.; Snoeck, R.; Schols, D.; De Clercq, E. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: Potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 1993, 37, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [Green Version]
- Pokorná, J.; Machala, L.; Rezáčová, P.; Konvalinka, J. Current and Novel Inhibitors of HIV Protease. Viruses 2009, 1, 1209–1239. [Google Scholar] [CrossRef] [Green Version]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef] [Green Version]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef]
- Engelman, K.D.; Engelman, A.N. Long-Acting Cabotegravir for HIV/AIDS Prophylaxis. Biochemistry 2021, 60, 1731–1740. [Google Scholar] [CrossRef]
- Snásel, J.; Rosenberg, I.; Paces, O.; Pichová, I. The strand transfer oligonucleotide inhibitors of HIV-integrase. J. Enzym. Inhib. Med. Chem. 2009, 24, 241–246. [Google Scholar] [CrossRef]
- Hikichi, Y.; Van Duyne, R.; Pham, P.; Groebner, J.L.; Wiegand, A.; Mellors, J.W.; Kearney, M.F.; Freed, E.O. Mechanistic Analysis of the Broad Antiretroviral Resistance Conferred by HIV-1 Envelope Glycoprotein Mutations. mBio 2021, 12, e03134-20. [Google Scholar] [CrossRef]
- De Andrade Arrais, C.R.; Lima, K.; Barreiros, M.; Rodrigues, J.K.F.; Sousa, N.P.S.; Costa, D.D.; Santos, F.; Pereira, G.F.M.; AI, E.S.V.; Barros, A.K.; et al. HIV-1 subtypes and drug resistance in children during antiretroviral therapy in Brazil. J. Med. Virol. 2021, 93, 4908–4914. [Google Scholar] [CrossRef]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Nka, A.D.; Teto, G.; Santoro, M.M.; Ngum Ndze, V.; Takou, D.; Dambaya, B.; Ngoufack Jagni Semengue, E.; Fabeni, L.; Perno, C.F.; Colizzi, V.; et al. HIV-1 Gag gene mutations, treatment response and drug resistance to protease inhibitors: A systematic review and meta-analysis protocol. PLoS ONE 2021, 16, e0253587. [Google Scholar] [CrossRef]
- Agniswamy, J.; Kneller, D.W.; Ghosh, A.K.; Weber, I.T. Novel HIV PR inhibitors with C4-substituted bis-THF and bis-fluoro-benzyl target the two active site mutations of highly drug resistant mutant PR(S17). Biochem. Biophys. Res. Commun. 2021, 566, 30–35. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Kozísek, M.; Henke, S.; Sasková, K.G.; Jacobs, G.B.; Schuch, A.; Buchholz, B.; Müller, V.; Kräusslich, H.G.; Rezácová, P.; Konvalinka, J.; et al. Mutations in HIV-1 gag and pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob. Agents Chemother. 2012, 56, 4320–4330. [Google Scholar] [CrossRef] [Green Version]
- Fun, A.; van Maarseveen, N.M.; Pokorná, J.; Maas, R.E.; Schipper, P.J.; Konvalinka, J.; Nijhuis, M. HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology 2011, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Sasková, K.G.; Kozísek, M.; Rezácová, P.; Brynda, J.; Yashina, T.; Kagan, R.M.; Konvalinka, J. Molecular characterization of clinical isolates of human immunodeficiency virus resistant to the protease inhibitor darunavir. J. Virol. 2009, 83, 8810–8818. [Google Scholar] [CrossRef] [Green Version]
- Mulato, A.; Acosta, R.; Chang, S.; Martin, R.; Yant, S.R.; Cihlar, T.; White, K. Simulating HIV Breakthrough and Resistance Development During Variable Adherence to Antiretroviral Treatment. J. Acquir. Immune Defic. Syndr. 2021, 86, 369–377. [Google Scholar] [CrossRef]
- Borghetti, A.; Ciccullo, A.; Lombardi, F.; Baldin, G.; Belmonti, S.; Prosperi, M.; Incardona, F.; Heger, E.; Borghi, V.; Sönnerborg, A.; et al. Transmitted drug resistance to NRTIs and risk of virological failure in naïve patients treated with integrase inhibitors. HIV Med. 2021, 22, 22–27. [Google Scholar] [CrossRef]
- Berríos-Caro, E.; Gifford, D.R.; Galla, T. Competition delays multi-drug resistance evolution during combination therapy. J. Theor. Biol. 2021, 509, 110524. [Google Scholar] [CrossRef]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Takamune, N.; Nirasawa, T.; Aoki, M.; Morishita, Y.; Das, D.; Koh, Y.; Ghosh, A.K.; Misumi, S.; Mitsuya, H. Dimerization of HIV-1 protease occurs through two steps relating to the mechanism of protease dimerization inhibition by darunavir. Proc. Natl. Acad. Sci. USA 2014, 111, 12234–12239. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Danish, M.L.; Aoki-Ogata, H.; Amano, M.; Ide, K.; Das, D.; Koh, Y.; Mitsuya, H. Loss of the protease dimerization inhibition activity of tipranavir (TPV) and its association with the acquisition of resistance to TPV by HIV-1. J. Virol. 2012, 86, 13384–13396. [Google Scholar] [CrossRef] [Green Version]
- Louis, J.M.; Aniana, A.; Weber, I.T.; Sayer, J.M. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9072–9077. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.; Soule, E.E.; Davidoff, K.S.; Daniels, S.I.; Naiman, N.E.; Yarchoan, R. Activity of human immunodeficiency virus type 1 protease inhibitors against the initial autocleavage in Gag-Pol polyprotein processing. Antimicrob. Agents Chemother. 2012, 56, 3620–3628. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Sayer, J.M.; Aniana, A.; Yu, X.; Weber, I.T.; Harrison, R.W.; Louis, J.M. Binding of Clinical Inhibitors to a Model Precursor of a Rationally Selected Multidrug Resistant HIV-1 Protease Is Significantly Weaker Than That to the Released Mature Enzyme. Biochemistry 2016, 55, 2390–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humpolíčková, J.; Weber, J.; Starková, J.; Mašínová, E.; Günterová, J.; Flaisigová, I.; Konvalinka, J.; Majerová, T. Inhibition of the precursor and mature forms of HIV-1 protease as a tool for drug evaluation. Sci. Rep. 2018, 8, 10438. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, N.K. Direct renin inhibition and the kidney. Nat. Rev. Nephrol. 2010, 6, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Chang, Y.-C.; Agniswamy, J.; Harrison, R.W.; Weber, I.T. Conformational variation of an extreme drug resistant mutant of HIV protease. J. Mol. Graph. Model. 2015, 62, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chang, Y.-C.E.; Louis, J.M.; Wang, Y.-F.; Harrison, R.W.; Weber, I.T. Structures of Darunavir-Resistant HIV-1 Protease Mutant Reveal Atypical Binding of Darunavir to Wide Open Flaps. ACS Chem. Biol. 2014, 9, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Tien, C.; LaBarbera, D.V.; Chen, C. Targeting HIV-1 Protease Autoprocessing for High-throughput Drug Discovery and Drug Resistance Assessment. Sci. Rep. 2019, 9, 301. [Google Scholar] [CrossRef]
- Aoki, M.; Das, D.; Hayashi, H.; Aoki-Ogata, H.; Takamatsu, Y.; Ghosh, A.K.; Mitsuya, H.; Prasad, V.R.; Shafer, R.; Kovari, L. Mechanism of Darunavir (DRV)’s High Genetic Barrier to HIV-1 Resistance: A Key V32I Substitution in Protease Rarely Occurs, but Once It Occurs, It Predisposes HIV-1 To Develop DRV Resistance. mBio 2018, 9, e02425-17. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Balasubramanian, S.; Senapati, S. Understanding the mechanism of HIV-1 protease inhibition by monoclonal antibodies. J. Mol. Graph. Model. 2021, 103, 107826. [Google Scholar] [CrossRef]
- Bowman, M.J.; Byrne, S.; Chmielewski, J. Switching between allosteric and dimerization inhibition of HIV-1 protease. Chem. Biol. 2005, 12, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Pietrucci, F.; Vargiu, A.V.; Kranjc, A. HIV-1 Protease Dimerization Dynamics Reveals a Transient Druggable Binding Pocket at the Interface. Sci. Rep. 2015, 5, 18555. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.; Matsumi, S.; Das, D.; Amano, M.; Davis, D.A.; Li, J.; Leschenko, S.; Baldridge, A.; Shioda, T.; Yarchoan, R.; et al. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 2007, 282, 28709–28720. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Poorman, R.A.; Maggiora, L.L.; Heinrikson, R.L.; Kézdy, F.J. Dissociative inhibition of dimeric enzymes. Kinetic characterization of the inhibition of HIV-1 protease by its COOH-terminal tetrapeptide. J. Biol. Chem. 1991, 266, 15591–15594. [Google Scholar] [CrossRef]
- Uhlíková, T.; Konvalinka, J.; Pichová, I.; Soucek, M.; Kräusslich, H.G.; Vondrásek, J. A modular approach to HIV-1 proteinase inhibitor design. Biochem. Biophys. Res. Commun. 1996, 222, 38–43. [Google Scholar] [CrossRef]
- Strisovsky, K.; Tessmer, U.; Langner, J.; Konvalinka, J.; Kräusslich, H.G. Systematic mutational analysis of the active-site threonine of HIV-1 proteinase: Rethinking the “fireman’s grip” hypothesis. Protein Sci. 2000, 9, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Ingr, M.; Uhlíková, T.; Strísovský, K.; Majerová, E.; Konvalinka, J. Kinetics of the dimerization of retroviral proteases: The “fireman’s grip” and dimerization. Protein Sci. 2003, 12, 2173–2182. [Google Scholar] [CrossRef]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Pettit, S.C.; Everitt, L.E.; Choudhury, S.; Dunn, B.M.; Kaplan, A.H. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol. 2004, 78, 8477–8485. [Google Scholar] [CrossRef] [Green Version]
- Pettit, S.C.; Clemente, J.C.; Jeung, J.A.; Dunn, B.M.; Kaplan, A.H. Ordered processing of the human immunodeficiency virus type 1 GagPol precursor is influenced by the context of the embedded viral protease. J. Virol. 2005, 79, 10601–10607. [Google Scholar] [CrossRef] [Green Version]
- Pettit, S.C.; Gulnik, S.; Everitt, L.; Kaplan, A.H. The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of GagPol cleavage. J. Virol. 2003, 77, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Tien, C.; Huang, L.; Watanabe, S.M.; Speidel, J.T.; Carter, C.A.; Chen, C. Context-dependent autoprocessing of human immunodeficiency virus type 1 protease precursors. PLoS ONE 2018, 13, e0191372. [Google Scholar] [CrossRef] [Green Version]
- Zábranský, A.; Andreánsky, M.; Hrusková-Heidingsfeldová, O.; Havlícek, V.; Hunter, E.; Ruml, T.; Pichová, I. Three active forms of aspartic proteinase from Mason-Pfizer monkey virus. Virology 1998, 245, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Veverka, V.; Bauerová, H.; Zábranský, A.; Lang, J.; Ruml, T.; Pichová, I.; Hrabal, R. Three-dimensional structure of a monomeric form of a retroviral protease. J. Mol. Biol. 2003, 333, 771–780. [Google Scholar] [CrossRef]
- Khatib, F.; DiMaio, F.; Cooper, S.; Kazmierczyk, M.; Gilski, M.; Krzywda, S.; Zabranska, H.; Pichova, I.; Thompson, J.; Popović, Z.; et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat. Struct. Mol. Biol. 2011, 18, 1175–1177. [Google Scholar] [CrossRef]
- Wosicki, S.; Kazmierczyk, M.; Gilski, M.; Zabranska, H.; Pichova, I.; Jaskolski, M. Crystal structures of inhibitor complexes of M-PMV protease with visible flap loops. Protein Sci. 2021, 30, 1258–1263. [Google Scholar] [CrossRef]
- Hrusková-Heidingsfeldová, O.; Andreansky, M.; Fábry, M.; Bláha, I.; Strop, P.; Hunter, E. Cloning, Bacterial Expression, and Characterization of the Mason-Pfizer Monkey Virus Proteinase. J. Biol. Chem. 1995, 270, 15053–15058. [Google Scholar] [CrossRef] [Green Version]
- Nijhuis, M.; van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; de Jong, D.; Chappey, C.; Goedegebuure, I.W.; et al. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007, 4, e36. [Google Scholar] [CrossRef]
- Xue, B.; Mizianty, M.J.; Kurgan, L.; Uversky, V.N. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell Mol. Life Sci. 2012, 69, 1211–1259. [Google Scholar] [CrossRef]
- Jochmans, D.; Anders, M.; Keuleers, I.; Smeulders, L.; Kräusslich, H.-G.; Kraus, G.; Müller, B. Selective killing of human immunodeficiency virus infected cells by non-nucleoside reverse transcriptase inhibitor-induced activation of HIV protease. Retrovirology 2010, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Sudo, S.; Haraguchi, H.; Hirai, Y.; Gatanaga, H.; Sakuragi, J.-I.; Momose, F.; Morikawa, Y. Efavirenz enhances HIV-1 gag processing at the plasma membrane through Gag-Pol dimerization. J. Virol. 2013, 87, 3348–3360. [Google Scholar] [CrossRef] [Green Version]
- Rumlová, M.; Křížová, I.; Keprová, A.; Hadravová, R.; Doležal, M.; Strohalmová, K.; Pichová, I.; Hájek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 37. [Google Scholar] [CrossRef]
- Buzon, M.J.; Erkizia, I.; Pou, C.; Minuesa, G.; Puertas, M.C.; Esteve, A.; Castello, A.; Santos, J.R.; Prado, J.G.; Izquierdo-Useros, N.; et al. A non-infectious cell-based phenotypic assay for the assessment of HIV-1 susceptibility to protease inhibitors. J. Antimicrob. Chemother. 2012, 67, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, K.; Uhlíková, T.; Konvalinka, J.; Masucci, M.G.; Dantuma, N.P. Cell-based fluorescence assay for human immunodeficiency virus type 1 protease activity. Antimicrob. Agents Chemother. 2001, 45, 2616–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majerová-Uhlíková, T.; Dantuma, N.P.; Lindsten, K.; Masucci, M.G.; Konvalinka, J. Non-infectious fluorimetric assay for phenotyping of drug-resistant HIV proteinase mutants. J. Clin. Virol. 2006, 36, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.H.; Swanstrom, R. Human immunodeficiency virus type 1 Gag proteins are processed in two cellular compartments. Proc. Natl. Acad. Sci. USA 1991, 88, 4528–4532. [Google Scholar] [CrossRef] [Green Version]
- Trinité, B.; Zhang, H.; Levy, D.N. NNRTI-induced HIV-1 protease-mediated cytotoxicity induces rapid death of CD4 T cells during productive infection and latency reversal. Retrovirology 2019, 16, 17. [Google Scholar] [CrossRef] [Green Version]
- Jurado, K.A.; Engelman, A. Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev. Mol. Med. 2013, 15, e14. [Google Scholar] [CrossRef] [Green Version]
- Kräusslich, H.G. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. USA 1991, 88, 3213–3217. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.Y.; Wang, S.M.; Huang, K.J.; Chiang, C.C.; Wang, C.T. Placement of leucine zipper motifs at the carboxyl terminus of HIV-1 protease significantly reduces virion production. PLoS ONE 2012, 7, e32845. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Wang, F.D.; Chen, Y.A.; Wang, C.T. Effects of human immunodeficiency virus type 1 transframe protein p6* mutations on viral protease-mediated Gag processing. J. Gen. Virol. 2006, 87, 2041–2046. [Google Scholar] [CrossRef]
- Zábranský, A.; Hadravová, R.; Stokrová, J.; Sakalian, M.; Pichová, I. Premature processing of mouse mammary tumor virus Gag polyprotein impairs intracellular capsid assembly. Virology 2009, 384, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Schur, F.K.; Hagen, W.J.; Rumlová, M.; Ruml, T.; Müller, B.; Kräusslich, H.G.; Briggs, J.A. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 2015, 517, 505–508. [Google Scholar] [CrossRef]
- Bohmová, K.; Hadravová, R.; Stokrová, J.; Tuma, R.; Ruml, T.; Pichová, I.; Rumlová, M. Effect of dimerizing domains and basic residues on in vitro and in vivo assembly of Mason-Pfizer monkey virus and human immunodeficiency virus. J. Virol. 2010, 84, 1977–1988. [Google Scholar] [CrossRef] [Green Version]
- Junková, P.; Pleskot, R.; Prchal, J.; Sýs, J.; Ruml, T. Differences and commonalities in plasma membrane recruitment of the two morphogenetically distinct retroviruses HIV-1 and MMTV. J. Biol. Chem. 2020, 295, 8819–8833. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; Ratcliff, A.N.; Bhat, M.; Alwarawrah, Y.; Hughes, P.; Arcos, J.; Loiselle, D.; Torrelles, J.B.; Funderburg, N.T.; Haystead, T.A.; et al. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology 2017, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Lindwasser, O.W.; Resh, M.D. Myristoylation as a target for inhibiting HIV assembly: Unsaturated fatty acids block viral budding. Proc. Natl. Acad. Sci. USA 2002, 99, 13037–13042. [Google Scholar] [CrossRef] [Green Version]
- Zell, R. Picornaviridae-the ever-growing virus family. Arch. Virol. 2018, 163, 299–317. [Google Scholar] [CrossRef]
- Sárkány, Z.; Polgár, L. The unusual catalytic triad of poliovirus protease 3C. Biochemistry 2003, 42, 516–522. [Google Scholar] [CrossRef]
- Horova, V.; Lyoo, H.; Różycki, B.; Chalupska, D.; Smola, M.; Humpolickova, J.; Strating, J.R.P.M.; van Kuppeveld, F.J.M.; Boura, E.; Klima, M. Convergent evolution in the mechanisms of ACBD3 recruitment to picornavirus replication sites. PLoS Pathog. 2019, 15, e1007962. [Google Scholar] [CrossRef] [Green Version]
- Parsley, T.B.; Cornell, C.T.; Semler, B.L. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J. Biol. Chem. 1999, 274, 12867–12876. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, L.L.; Wass, A.B.; Gohara, D.W.; Pathak, H.B.; Arnold, J.J.; Filman, D.J.; Cameron, C.E.; Hogle, J.M. Crystal structure of poliovirus 3CD protein: Virally encoded protease and precursor to the RNA-dependent RNA polymerase. J. Virol. 2007, 81, 3583–3596. [Google Scholar] [CrossRef] [Green Version]
- Winston, D.S.; Boehr, D.D. The Picornavirus Precursor 3CD Has Different Conformational Dynamics Compared to 3C(pro) and 3D(pol) in Functionally Relevant Regions. Viruses 2021, 13, 442. [Google Scholar] [CrossRef]
- Spear, A.; Ogram, S.A.; Morasco, B.J.; Smerage, L.E.; Flanegan, J.B. Viral precursor protein P3 and its processed products perform discrete and essential functions in the poliovirus RNA replication complex. Virology 2015, 485, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.M.; Moustafa, I.M.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Long-Range Communication between Different Functional Sites in the Picornaviral 3C Protein. Structure 2016, 24, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Meng, B.; Lan, K.; Xie, J.; Lerner, R.A.; Wilson, I.A.; Yang, B. Inhibitory antibodies identify unique sites of therapeutic vulnerability in rhinovirus and other enteroviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 13499–13508. [Google Scholar] [CrossRef]
- Rahnefeld, A.; Klingel, K.; Schuermann, A.; Diny, N.L.; Althof, N.; Lindner, A.; Bleienheuft, P.; Savvatis, K.; Respondek, D.; Opitz, E.; et al. Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation 2014, 130, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, E.M.; James, M.N.G. The 3C proteinases of picornaviruses. In Proteases as Targets for Therapy; von der Helm, K., Korant, B.D., Cheronis, J.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 117–143. [Google Scholar]
- Green, K.Y.; Kaufman, S.S.; Nagata, B.M.; Chaimongkol, N.; Kim, D.Y.; Levenson, E.A.; Tin, C.M.; Yardley, A.B.; Johnson, J.A.; Barletta, A.B.F.; et al. Human norovirus targets enteroendocrine epithelial cells in the small intestine. Nat. Commun. 2020, 11, 2759. [Google Scholar] [CrossRef]
- Muzzarelli, K.M.; Kuiper, B.; Spellmon, N.; Brunzelle, J.; Hackett, J.; Amblard, F.; Zhou, S.; Liu, P.; Kovari, I.A.; Yang, Z.; et al. Structural and Antiviral Studies of the Human Norovirus GII.4 Protease. Biochemistry 2019, 58, 900–907. [Google Scholar] [CrossRef]
- Viskovska, M.A.; Zhao, B.; Shanker, S.; Choi, J.-M.; Deng, L.; Song, Y.; Palzkill, T.; Hu, L.; Estes, M.K.; Prasad, B.V.V.; et al. GII.4 Norovirus Protease Shows pH-Sensitive Proteolysis with a Unique Arg-His Pairing in the Catalytic Site. J. Virol. 2019, 93, e01479-18. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.O.; Kim, Y.; Lovell, S.; Rathnayake, A.D.; Groutas, W.C. Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors. Viruses 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Abu Bakar, F.; Ng, L.F.P. Nonstructural Proteins of Alphavirus—Potential Targets for Drug Development. Viruses 2018, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.Y.; Firth, A.E.; Atkins, J.F. Frameshifting in alphaviruses: A diversity of 3’ stimulatory structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Potužník, J.F.; Cahová, H. It’s the Little Things (in Viral RNA). mBio 2020, 11, e02131-20. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J.; Hardy, W.R.; Shirako, Y.; Strauss, J.H. Cleavage-site preferences of Sindbis virus polyproteins containing the non-structural proteinase. Evidence for temporal regulation of polyprotein processing in vivo. EMBO J. 1990, 9, 2631–2638. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2021, 589, 615–619. [Google Scholar] [CrossRef]
- Hellström, K.; Kallio, K.; Utt, A.; Quirin, T.; Jokitalo, E.; Merits, A.; Ahola, T.; Simon, A.E. Partially Uncleaved Alphavirus Replicase Forms Spherule Structures in the Presence and Absence of RNA Template. J. Virol. 2017, 91, e00787-17. [Google Scholar] [CrossRef] [Green Version]
- Shirako, Y.; Strauss, J.H. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994, 68, 1874–1885. [Google Scholar] [CrossRef] [Green Version]
- Lemm, J.A.; Rümenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534–16539. [Google Scholar] [CrossRef] [Green Version]
- Lulla, A.; Lulla, V.; Tints, K.; Ahola, T.; Merits, A. Molecular determinants of substrate specificity for Semliki Forest virus nonstructural protease. J. Virol. 2006, 80, 5413–5422. [Google Scholar] [CrossRef] [Green Version]
- Lulla, V.; Karo-Astover, L.; Rausalu, K.; Saul, S.; Merits, A.; Lulla, A. Timeliness of Proteolytic Events Is Prerequisite for Efficient Functioning of the Alphaviral Replicase. J. Virol. 2018, 92, e00151-18. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Castle, E.; Nowak, T.; Leidner, U.; Wengler, G.; Wengler, G. Sequence analysis of the viral core protein and the membrane-associated proteins V1 and NV2 of the flavivirus west nile virus and of the genome sequence for these proteins. Virology 1985, 145, 227–236. [Google Scholar] [CrossRef]
- Nowak, T.; Färber, P.M.; Wengler, G.; Wengler, G. Analyses of the terminal sequences of west nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology 1989, 169, 365–376. [Google Scholar] [CrossRef]
- Falgout, B.; Pethel, M.; Zhang, Y.M.; Lai, C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991, 65, 2467–2475. [Google Scholar] [CrossRef] [Green Version]
- Amberg, S.M.; Nestorowicz, A.; McCourt, D.W.; Rice, C.M. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: In vitro and in vivo studies. J. Virol. 1994, 68, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Rana, J.; Burrone, O.R. DENV2 Pseudoviral Particles with Unprocessed Capsid Protein Are Assembled and Infectious. Viruses 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Kurz, M.; Stefan, N.; Zhu, J.; Skern, T. NS2B/3 proteolysis at the C-prM junction of the tick-borne encephalitis virus polyprotein is highly membrane dependent. Virus Res. 2012, 168, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.-M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the Immature Dengue Virus at Low pH Primes Proteolytic Maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef]
- Plevka, P.; Battisti, A.J.; Sheng, J.; Rossmann, M.G. Mechanism for maturation-related reorganization of flavivirus glycoproteins. J. Struct. Biol. 2014, 185, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Rice, C.M. The hepatitis C virus NS3 serine proteinase and NS4A cofactor: Establishment of a cell-free trans-processing assay. Proc. Natl. Acad. Sci. USA 1995, 92, 7622–7626. [Google Scholar] [CrossRef] [Green Version]
- Clark, V.C.; Peter, J.A.; Nelson, D.R. New therapeutic strategies in HCV: Second-generation protease inhibitors. Liver Int. 2013, 33 Suppl 1, 80–84. [Google Scholar] [CrossRef]
- Majerová, T.; Novotný, P.; Krýsová, E.; Konvalinka, J. Exploiting the unique features of Zika and Dengue proteases for inhibitor design. Biochimie 2019, 166, 132–141. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Structure and Dynamics of Zika Virus Protease and Its Insights into Inhibitor Design. Biomedicines 2021, 9, 1044. [Google Scholar] [CrossRef]
- Gupta, G.; Lim, L.; Song, J. NMR and MD Studies Reveal That the Isolated Dengue NS3 Protease Is an Intrinsically Disordered Chymotrypsin Fold Which Absolutely Requests NS2B for Correct Folding and Functional Dynamics. PLoS ONE 2015, 10, e0134823. [Google Scholar] [CrossRef]
- Yusof, R.; Clum, S.; Wetzel, M.; Murthy, H.M.K.; Padmanabhan, R. Purified NS2B/NS3 Serine Protease of Dengue Virus Type 2 Exhibits Cofactor NS2B Dependence for Cleavage of Substrates with Dibasic Amino Acids. J. Biol. Chem. 2000, 275, 9963–9969. [Google Scholar] [CrossRef] [Green Version]
- Brinkworth, R.I.; Fairlie, D.P.; Leung, D.; Young, P.R. Homology model of the dengue 2 virus NS3 protease: Putative interactions with both substrate and NS2B cofactor. J. Gen. Virol. 1999, 80, 1167–1177. [Google Scholar] [CrossRef]
- Leung, D.; Schroder, K.; White, H.; Fang, N.-X.; Stoermer, M.J.; Abbenante, G.; Martin, J.L.; Young, P.R.; Fairlie, D.P. Activity of Recombinant Dengue 2 Virus NS3 Protease in the Presence of a Truncated NS2B Co-factor, Small Peptide Substrates, and Inhibitors. J. Biol. Chem. 2001, 276, 45762–45771. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [Green Version]
- Behnam, M.A.M.; Klein, C.D.P. Conformational selection in the flaviviral NS2B-NS3 protease. Biochimie 2020, 174, 117–125. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Milani, M.; Bollati, M.; Selisko, B.; Peyrane, F.; Pandini, V.; Sorrentino, G.; Canard, B.; Konarev, P.V.; Svergun, D.I.; et al. Crystal Structure and Activity of Kunjin Virus NS3 Helicase; Protease and Helicase Domain Assembly in the Full Length NS3 Protein. J. Mol. Biol. 2007, 372, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue Virus Helicase/Nucleoside Triphosphatase Catalytic Domain at a Resolution of 2.4 A. J. Virol. 2005, 79, 10278–10288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Wei, N.; Doan, D.N.; Paradkar, P.N.; Chong, Y.; Davidson, A.D.; Kotaka, M.; Lescar, J.; Vasudevan, S.G. Flexibility between the protease and helicase domains of the dengue virus NS3 protein conferred by the linker region and its functional implications. J. Biol. Chem. 2010, 285, 18817–18827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Huo, T.; Lin, Y.L.; Nie, S.; Wu, F.; Hua, Y.; Wu, J.; Kneubehl, A.R.; Vogt, M.B.; Rico-Hesse, R.; et al. Discovery, X-ray Crystallography and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Am. Chem. Soc. 2019, 141, 6832–6836. [Google Scholar] [CrossRef]
- Teo, K.F.; Wright, P.J. Internal proteolysis of the NS3 protein specified by dengue virus 2. J. Gen. Virol. 1997, 78, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Kümmerer, B.M.; Rice, C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002, 76, 4773–4784. [Google Scholar] [CrossRef] [Green Version]
- Constant, D.A.; Mateo, R.; Nagamine, C.M.; Kirkegaard, K. Targeting intramolecular proteinase NS2B/3 cleavages for trans-dominant inhibition of dengue virus. Proc. Natl. Acad. Sci. USA 2018, 115, 10136–10141. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.J.H.; Lee, R.C.H.; Ang, M.J.Y.; Wang, W.-L.; Lim, H.A.; Wee, J.L.K.; Joy, J.; Hill, J.; Brian Chia, C.S. Antiviral activities of 15 dengue NS2B-NS3 protease inhibitors using a human cell-based viral quantification assay. Antivir. Res. 2015, 118, 68–74. [Google Scholar] [CrossRef]
- Tomlinson, S.M.; Watowich, S.J. Anthracene-based inhibitors of dengue virus NS2B–NS3 protease. Antivir. Res. 2011, 89, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, C.; Zogali, V.; Chan, K.W.K.; Kiousis, D.; Gwee, C.P.; Wang, S.; Lescar, J.; Luo, D.; von Itzstein, M.; Matsoukas, M.-T.; et al. Amidoxime prodrugs convert to potent cell-active multimodal inhibitors of the dengue virus protease. Eur. J. Med. Chem. 2021, 224, 113695. [Google Scholar] [CrossRef]
- Richter, M.; Leuthold, M.M.; Graf, D.; Bartenschlager, R.; Klein, C.D. Prodrug Activation by a Viral Protease: Evaluating Combretastatin Peptide Hybrids to Selectively Target Infected Cells. ACS Med. Chem. Lett. 2019, 10, 1115–1121. [Google Scholar] [CrossRef]
- Luo, D.; Xu, T.; Hunke, C.; Grüber, G.; Vasudevan, S.G.; Lescar, J. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 2008, 82, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Poon, R.W.S.; Wong, B.H.L.; Tsoi, H.-W.; Yip, B.C.K.; Huang, Y.; Chan, K.-H.; Yuen, K.-Y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Liya, G.; Yuguang, W.; Jian, L.; Huaiping, Y.; Xue, H.; Jianwei, H.; Jiaju, M.; Youran, L.; Chen, M.; Yiqing, J. Studies on viral pneumonia related to novel coronavirus SARS-CoV-2, SARS-CoV, and MERS-CoV: A literature review. APMIS 2020, 128, 423–432. [Google Scholar] [CrossRef]
- Spaan, W.; Cavanagh, D.; Horzinek, M.C. Coronaviruses: Structure and Genome Expression. J. Gen. Virol. 1988, 69, 2939–2952. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-h.; et al. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Ratia, K.; Saikatendu, K.S.; Santarsiero, B.D.; Barretto, N.; Baker, S.C.; Stevens, R.C.; Mesecar, A.D. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [Google Scholar] [CrossRef] [Green Version]
- Ziebuhr, J.; Herold, J.; Siddell, S.G. Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J. Virol. 1995, 69, 4331–4338. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef]
- Grum-Tokars, V.; Ratia, K.; Begaye, A.; Baker, S.C.; Mesecar, A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: Recommendations for standardized assays for drug discovery. Virus Res. 2008, 133, 63–73. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, N.; Xue, F.; Kang, X.; Ren, X.; Chen, J.; Jin, C.; Lou, Z.; Xia, B. Three-dimensional domain swapping as a mechanism to lock the active conformation in a super-active octamer of SARS-CoV main protease. Protein Cell 2010, 1, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wei, P.; Huang, C.; Tan, L.; Liu, Y.; Lai, L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006, 281, 13894–13898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Jaskolski, M.; Dauter, Z.; Shabalin, I.G.; Gilski, M.; Brzezinski, D.; Kowiel, M.; Rupp, B.; Wlodawer, A. Crystallographic models of SARS-CoV-2 3CLpro: In-depth assessment of structure quality and validation. IUCrJ 2021, 8, 238–256. [Google Scholar] [CrossRef] [PubMed]
- Jaffrelot Inizan, T.; Célerse, F.; Adjoua, O.; El Ahdab, D.; Jolly, L.H.; Liu, C.; Ren, P.; Montes, M.; Lagarde, N.; Lagardère, L.; et al. High-resolution mining of the SARS-CoV-2 main protease conformational space: Supercomputer-driven unsupervised adaptive sampling. Chem. Sci. 2021, 12, 4889–4907. [Google Scholar] [CrossRef] [PubMed]
- Barrila, J.; Bacha, U.; Freire, E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 2006, 45, 14908–14916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, M.F.; Kuo, C.J.; Chang, K.T.; Chang, H.C.; Chou, C.C.; Ko, T.P.; Shr, H.L.; Chang, G.G.; Wang, A.H.; Liang, P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, T.; Kim, Y.-T.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J. 2013, 280, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Jonas, F.; Shen, C.; Hilgenfeld, R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 2010, 1, 59–74. [Google Scholar] [CrossRef]
- Noske, G.D.; Nakamura, A.M.; Gawriljuk, V.O.; Fernandes, R.S.; Lima, G.M.A.; Rosa, H.V.D.; Pereira, H.D.; Zeri, A.C.M.; Nascimento, A.F.Z.; Freire, M.C.L.C.; et al. A Crystallographic Snapshot of SARS-CoV-2 Main Protease Maturation Process. J. Mol. Biol. 2021, 433, 167118. [Google Scholar] [CrossRef]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J. Gen. Virol. 2021, 102, 1558. [Google Scholar] [CrossRef]
- Lee, J.; Worrall, L.J.; Vuckovic, M.; Rosell, F.I.; Gentile, F.; Ton, A.-T.; Caveney, N.A.; Ban, F.; Cherkasov, A.; Paetzel, M.; et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020, 11, 5877. [Google Scholar] [CrossRef]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, Purification, and Substrate Specificity of Severe Acute Respiratory Syndrome Coronavirus 3C-like Proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-Like Protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-C.; Chang, G.-G.; Chou, C.-Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Muhaxhiri, Z.; Deng, L.; Shanker, S.; Sankaran, B.; Estes, M.K.; Palzkill, T.; Song, Y.; Prasad, B.V.V. Structural basis of substrate specificity and protease inhibition in Norwalk virus. J. Virol. 2013, 87, 4281–4292. [Google Scholar] [CrossRef] [Green Version]
- Ingr, M.; Lange, R.; Halabalová, V.; Yehya, A.; Hrnčiřík, J.; Chevalier-Lucia, D.; Palmade, L.; Blayo, C.; Konvalinka, J.; Dumay, E. Inhibitor and substrate binding induced stability of HIV-1 protease against sequential dissociation and unfolding revealed by high pressure spectroscopy and kinetics. PLoS ONE 2015, 10, e0119099. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Lim, L.; Gupta, G.; Roy, A.; Kang, J.; Srivastava, S.; Shi, J.; Song, J. Structurally- and dynamically-driven allostery of the chymotrypsin-like proteases of SARS, Dengue and Zika viruses. Prog. Biophys. Mol. Biol. 2019, 143, 52–66. [Google Scholar] [CrossRef]
- Kokkonen, P.; Slanska, M.; Dockalova, V.; Pinto, G.P.; Sánchez-Carnerero, E.M.; Damborsky, J.; Klán, P.; Prokop, Z.; Bednar, D. The impact of tunnel mutations on enzymatic catalysis depends on the tunnel-substrate complementarity and the rate-limiting step. Comput. Struct. Biotechnol. J. 2020, 18, 805–813. [Google Scholar] [CrossRef]
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 2020, 12, eabc5332. [Google Scholar] [CrossRef]
- Kim, Y.; Shivanna, V.; Narayanan, S.; Prior, A.M.; Weerasekara, S.; Hua, D.H.; Kankanamalage, A.C.G.; Groutas, W.C.; Chang, K.-O. Broad-spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses. J. Virol. 2015, 89, 4942–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.S.; Hui, K.P.Y.; Lai, H.M.; He, X.; Khan, K.S.; Kaur, S.; Huang, J.; Li, Z.; Chan, A.K.N.; Cheung, H.H.; et al. Simeprevir Potently Suppresses SARS-CoV-2 Replication and Synergizes with Remdesivir. ACS Cent. Sci. 2021, 7, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- Tian, D.; Liu, Y.; Liang, C.; Xin, L.; Xie, X.; Zhang, D.; Wan, M.; Li, H.; Fu, X.; Liu, H.; et al. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother. 2021, 137, 111313. [Google Scholar] [CrossRef]
- Yang, H.; Yang, J. A review of the latest research on M(pro) targeting SARS-COV inhibitors. RSC Med. Chem. 2021, 12, 1026–1036. [Google Scholar] [CrossRef]
- El-Baba, T.J.; Lutomski, C.A.; Kantsadi, A.L.; Malla, T.R.; John, T.; Mikhailov, V.; Bolla, J.R.; Schofield, C.J.; Zitzmann, N.; Vakonakis, I.; et al. Allosteric Inhibition of the SARS-CoV-2 Main Protease: Insights from Mass Spectrometry Based Assays. Angew. Chem. Int. Ed. Engl. 2020, 59, 23544–23548. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. [Google Scholar] [CrossRef]
- Rizzuti, B.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Vega, S.; Grande, F.; Conforti, F.; Abian, O.; Velazquez-Campoy, A. Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds. Pharmaceuticals 2021, 14, 892. [Google Scholar] [CrossRef]
- Peñalver, L.; Schmid, P.; Szamosvári, D.; Schildknecht, S.; Globisch, C.; Sawade, K.; Peter, C.; Böttcher, T.A. Ligand Selection Strategy Identifies Chemical Probes Targeting the Proteases of SARS-CoV-2. Angew. Chem. Int. Ed. 2021, 60, 6799–6806. [Google Scholar] [CrossRef]
- Kneller, D.W.; Galanie, S.; Phillips, G.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Malleability of the SARS-CoV-2 3CL M(pro) Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure 2020, 28, 1313–1320. [Google Scholar] [CrossRef]
- Behnam, M.A.M. Protein structural heterogeneity: A hypothesis for the basis of proteolytic recognition by the main protease of SARS-CoV and SARS-CoV-2. Biochimie 2021, 182, 177–184. [Google Scholar] [CrossRef]
- Mattei, S.; Anders, M.; Konvalinka, J.; Kräusslich, H.-G.; Briggs, J.A.G.; Müller, B. Induced maturation of human immunodeficiency virus. J. Virol. 2014, 88, 13722–13731. [Google Scholar] [CrossRef] [Green Version]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef] [Green Version]
- Kanjanahaluethai, A.; Baker, S.C. Identification of mouse hepatitis virus papain-like proteinase 2 activity. J. Virol. 2000, 74, 7911–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, S.G.; Sawicki, D.L.; Younker, D.; Meyer, Y.; Thiel, V.; Stokes, H.; Siddell, S.G. Functional and Genetic Analysis of Coronavirus Replicase-Transcriptase Proteins. PLoS Pathog. 2005, 1, e39. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, Y.; Ratia, K.; Mesecar, A.D.; Wilkinson, K.D.; Baker, S.C. Proteolytic Processing and Deubiquitinating Activity of Papain-Like Proteases of Human Coronavirus NL63. J. Virol. 2007, 81, 6007–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wu, G. Spatial and temporal roles of SARS-CoV PLpro—A snapshot. FASEB J. 2021, 35, e21197. [Google Scholar] [CrossRef]
- Cihlova, B.; Huskova, A.; Böserle, J.; Nencka, R.; Boura, E.; Silhan, J. High-Throughput Fluorescent Assay for Inhibitor Screening of Proteases from RNA Viruses. Molecules 2021, 26, 3792. [Google Scholar] [CrossRef]
- Lin, M.H.; Moses, D.C.; Hsieh, C.H.; Cheng, S.C.; Chen, Y.H.; Sun, C.Y.; Chou, C.Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir. Res. 2018, 150, 155–163. [Google Scholar] [CrossRef]
- Ara, A.; Kadoya, R.; Ishimura, H.; Shimamura, K.; Sylte, I.; Kurita, N. Specific interactions between zinc metalloproteinase anditsinhibitors: Ab initio fragment molecular orbital calculations. J. Mol. Graph. Model. 2017, 75, 277–286. [Google Scholar] [CrossRef]
- Armstrong, L.A.; Lange, S.M.; Dee Cesare, V.; Matthews, S.P.; Nirujogi, R.S.; Cole, I.; Hope, A.; Cunningham, F.; Toth, R.; Mukherjee, R.; et al. Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition bynanobodies. PLoS ONE 2021, 16, e0253364. [Google Scholar] [CrossRef]
Figure 1.
Herpesviruses: Two fusion proteins expressed from RNAs identical at the 3’-terminus, namely the protease precursor (pPR) embedding assemblin and the assembly protein precursor (pAP). The major capsid protein binding domain (blue) ensures the formation of a complex with the capsid protein. This complex is recruited into the cell nucleus via its nuclear localization sequence (red). Association of capsid proteins mediates the dimerization of assemblin, leading to its autoactivation. The scaffold protein is proteolytically processed at the M and R sites. The intra-assemblin processing sites I and C have regulatory roles.
Figure 1.
Herpesviruses: Two fusion proteins expressed from RNAs identical at the 3’-terminus, namely the protease precursor (pPR) embedding assemblin and the assembly protein precursor (pAP). The major capsid protein binding domain (blue) ensures the formation of a complex with the capsid protein. This complex is recruited into the cell nucleus via its nuclear localization sequence (red). Association of capsid proteins mediates the dimerization of assemblin, leading to its autoactivation. The scaffold protein is proteolytically processed at the M and R sites. The intra-assemblin processing sites I and C have regulatory roles.

Figure 2.
Adenoviruses are characterized by the activation of adenoviral protease (adenain); the activity of adenain (AVP) is stimulated by DNA and by the pVIc peptide released from the pre-pVI protein. Peptide pVIc forms a disulfide bond with adenain and, as a result, increases its proteolytic activity further.
Figure 2.
Adenoviruses are characterized by the activation of adenoviral protease (adenain); the activity of adenain (AVP) is stimulated by DNA and by the pVIc peptide released from the pre-pVI protein. Peptide pVIc forms a disulfide bond with adenain and, as a result, increases its proteolytic activity further.

Figure 3.
FDA-approved inhibitors of the HIV protease.

Figure 4.
Retroviruses: Gag and Gag-Pol polyprotein of HIV-1 with its N-terminal myristoyl membrane anchor. The ratio of the Gag and Gag-Pol molecules is precisely regulated by the frequency of frameshift events. The arrows indicate the initial cis-cleavage sites.
Figure 4.
Retroviruses: Gag and Gag-Pol polyprotein of HIV-1 with its N-terminal myristoyl membrane anchor. The ratio of the Gag and Gag-Pol molecules is precisely regulated by the frequency of frameshift events. The arrows indicate the initial cis-cleavage sites.

Figure 5.
Compounds binding precursors of viral proteases: efavirenz binds to the Gag-Pol precursor, leading to the premature activation of the HIV protease, whereas ARDP0006 and the carbazole derivative SP-471 block intrahelicase cis-cleavage in Dengue NS3.
Figure 5.
Compounds binding precursors of viral proteases: efavirenz binds to the Gag-Pol precursor, leading to the premature activation of the HIV protease, whereas ARDP0006 and the carbazole derivative SP-471 block intrahelicase cis-cleavage in Dengue NS3.

Figure 6.
FDA-approved inhibitors of the HCV protease.

Figure 7.
Overall structure of the Dengue NS2B-NS3 heterodimer, which harbors the protease needed for releasing viral proteins from the precursor (A). Close-up of the linker connecting the protease and helicase domains of NS3 (B). The protease domain is shown in green, the helicase domain is colored in blue, and the activating peptide of NS2B is yellow. The ten amino acids linker, connecting the two enzymatically independent domains of NS3, is shown in pink (carbon backbone). Within the linker, oxygen and nitrogen atoms are shown in red and blue, respectively. The PDB accession number of the structure shown is 2VBC [193].
Figure 7.
Overall structure of the Dengue NS2B-NS3 heterodimer, which harbors the protease needed for releasing viral proteins from the precursor (A). Close-up of the linker connecting the protease and helicase domains of NS3 (B). The protease domain is shown in green, the helicase domain is colored in blue, and the activating peptide of NS2B is yellow. The ten amino acids linker, connecting the two enzymatically independent domains of NS3, is shown in pink (carbon backbone). Within the linker, oxygen and nitrogen atoms are shown in red and blue, respectively. The PDB accession number of the structure shown is 2VBC [193].

Figure 8.
Polyprotein precursor pp1ab of SARS-CoV-2. Precursors of coronaviral proteases are anchored to the membrane of the endoplasmic reticulum by adjacent transmembrane domains of neighboring proteins.
Figure 8.
Polyprotein precursor pp1ab of SARS-CoV-2. Precursors of coronaviral proteases are anchored to the membrane of the endoplasmic reticulum by adjacent transmembrane domains of neighboring proteins.

Figure 9.
Inhibitors of SARS-CoV-2 Mpro in clinical trials (A) and inhibitors of the Mpro precursor (B).
Figure 9.
Inhibitors of SARS-CoV-2 Mpro in clinical trials (A) and inhibitors of the Mpro precursor (B).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Majerová, T.; Novotný, P. Precursors of Viral Proteases as Distinct Drug Targets. Viruses 2021, 13, 1981. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101981
AMA Style
Majerová T, Novotný P. Precursors of Viral Proteases as Distinct Drug Targets. Viruses. 2021; 13(10):1981. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101981
Chicago/Turabian StyleMajerová, Taťána, and Pavel Novotný. 2021. "Precursors of Viral Proteases as Distinct Drug Targets" Viruses 13, no. 10: 1981. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101981
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.