RAC1B Regulation of TGFB1 Reveals an Unexpected Role of Autocrine TGFβ1 in the Suppression of Cell Motility

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
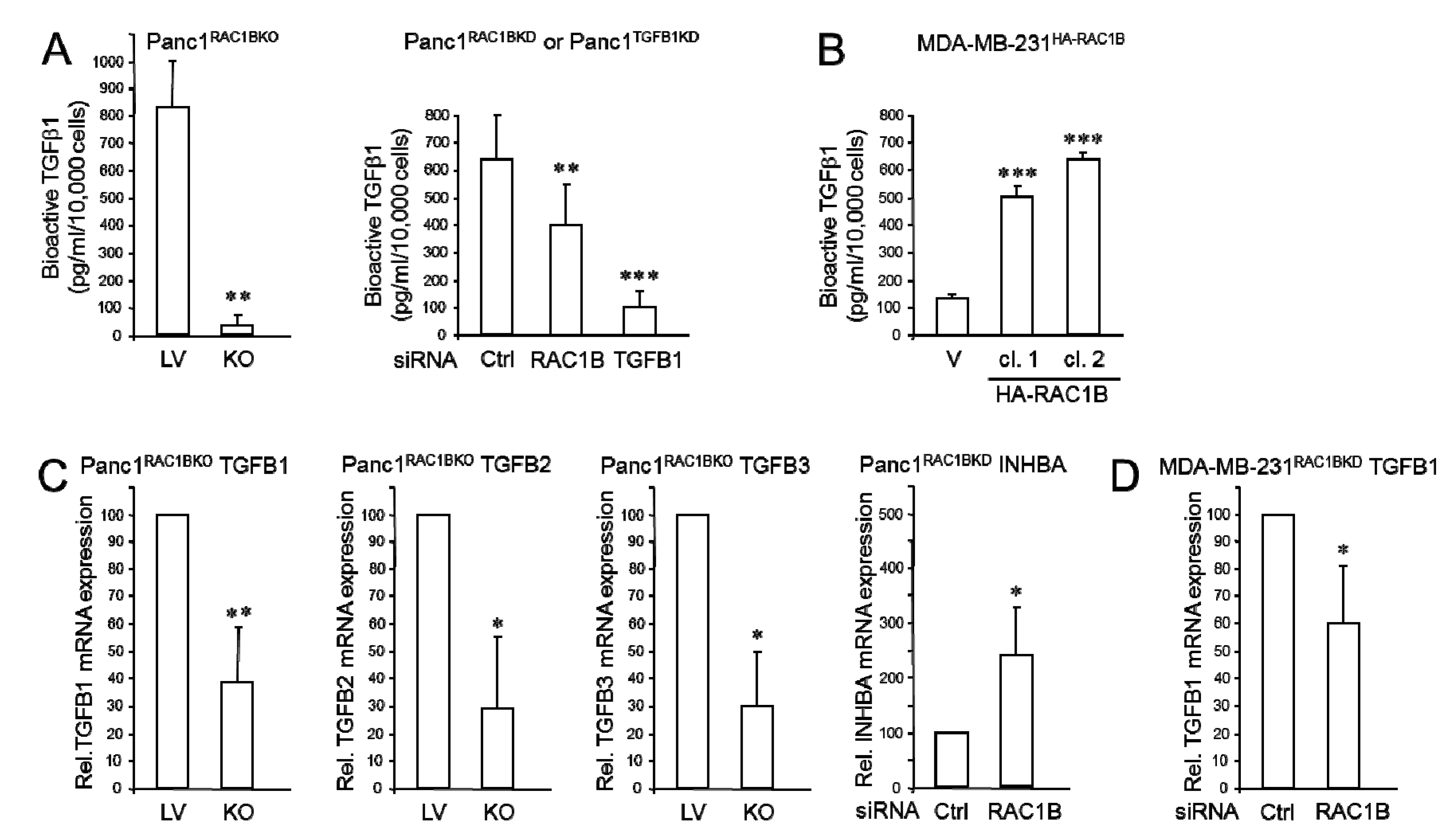
2.1. RAC1B Promotes Expression and Secretion of TGFβ1
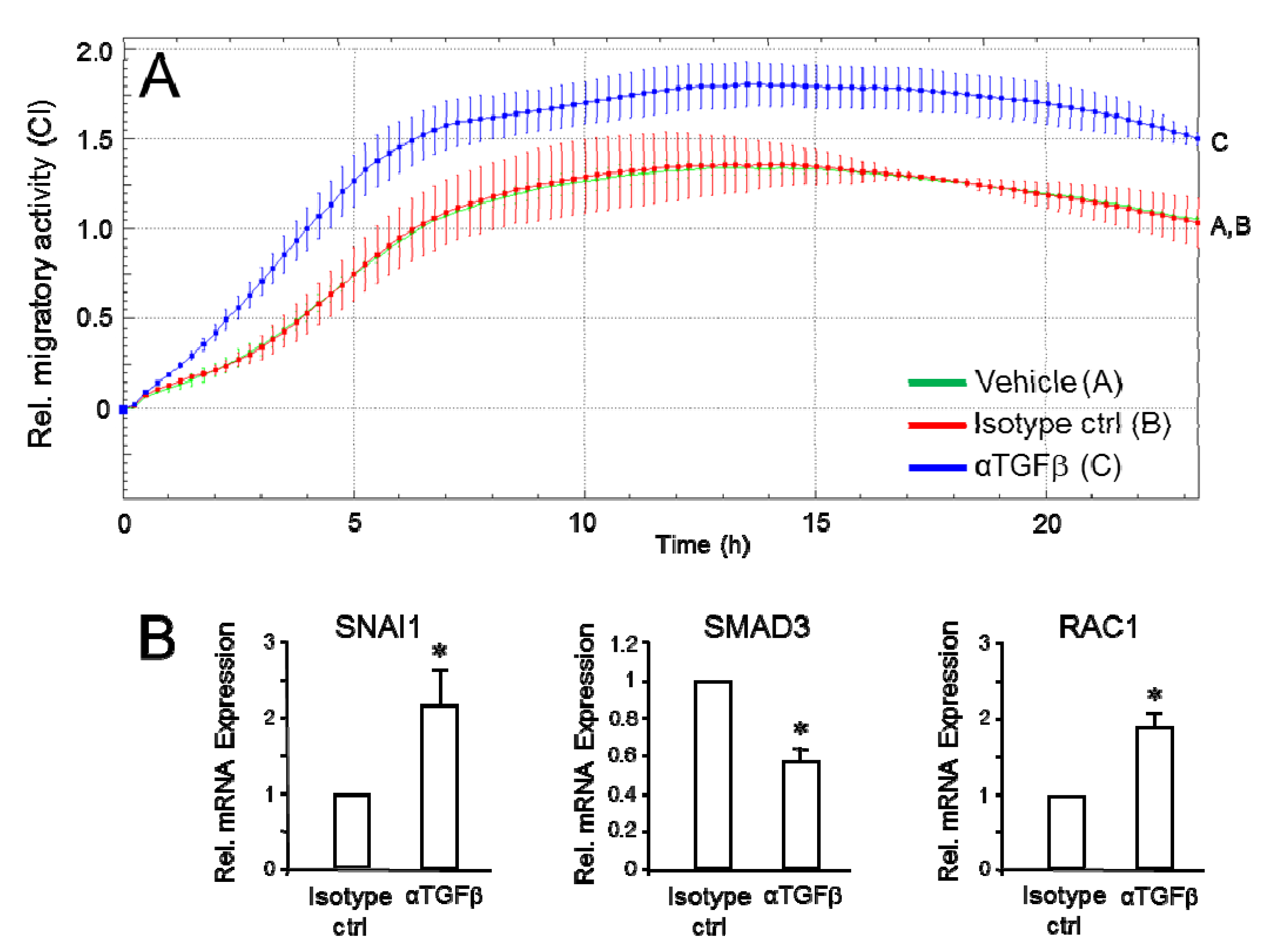
2.2. Effect of Neutralizing Autocrine TGFβ on Cell Migration and Gene Expression
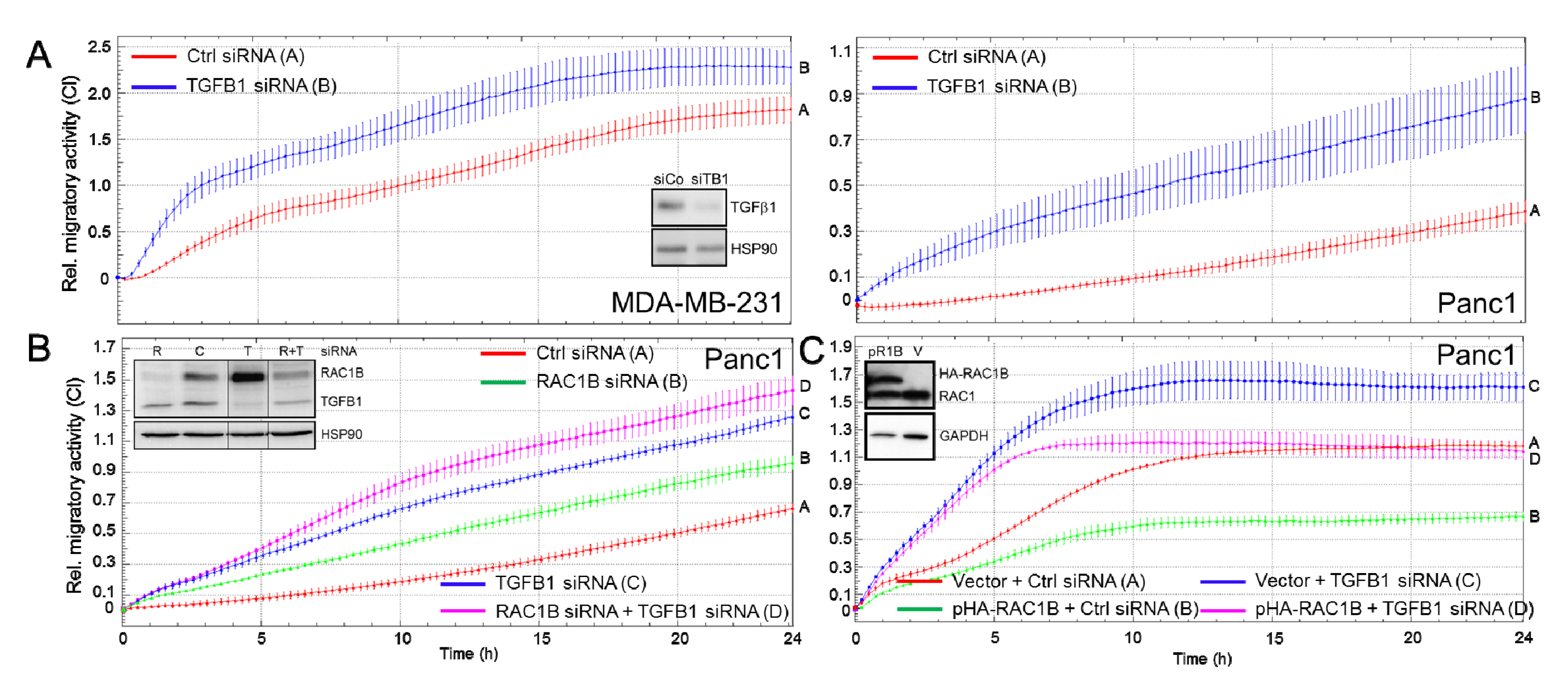
2.3. Endogenous TGFB1 Mimics the Suppressive Effect of RAC1B on Cell Motility
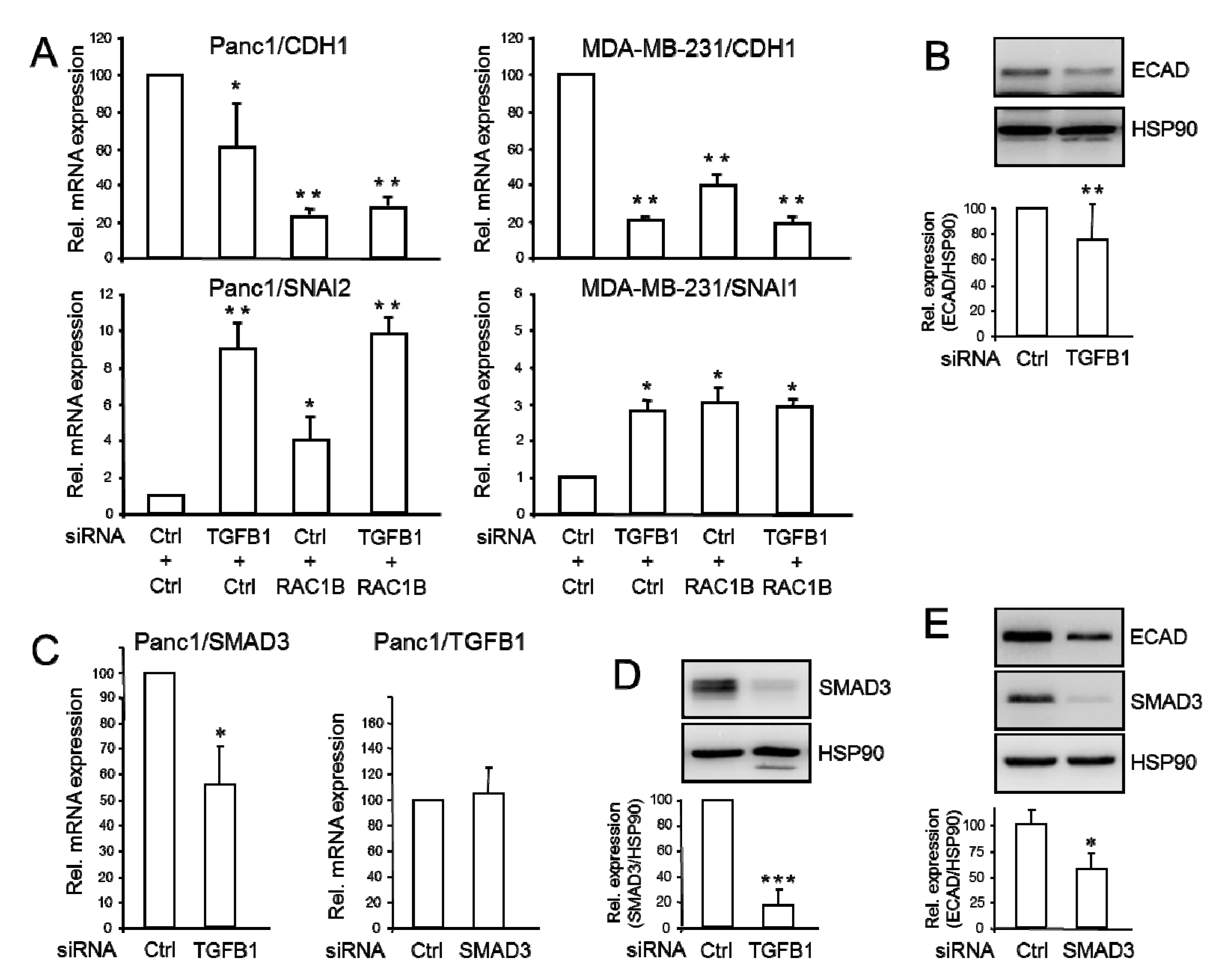
2.4. Endogenous TGFB1 Promotes CDH1 and SMAD3, and Suppresses SNAI1 Expression
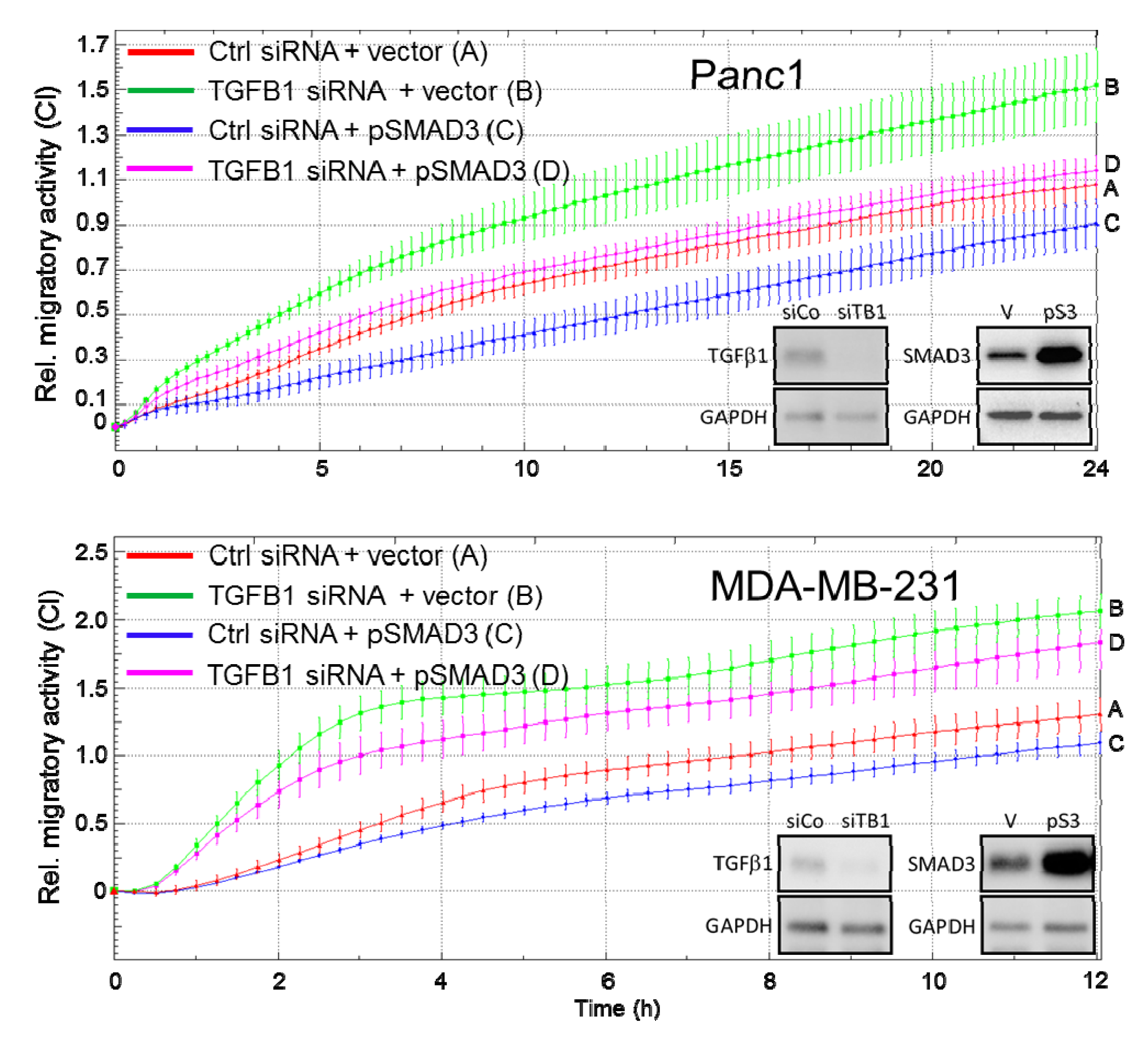
2.5. Ectopic Expression of SMAD3 Rescues Cells from the TGFB1 KD-Induced Increase in Cell Migration
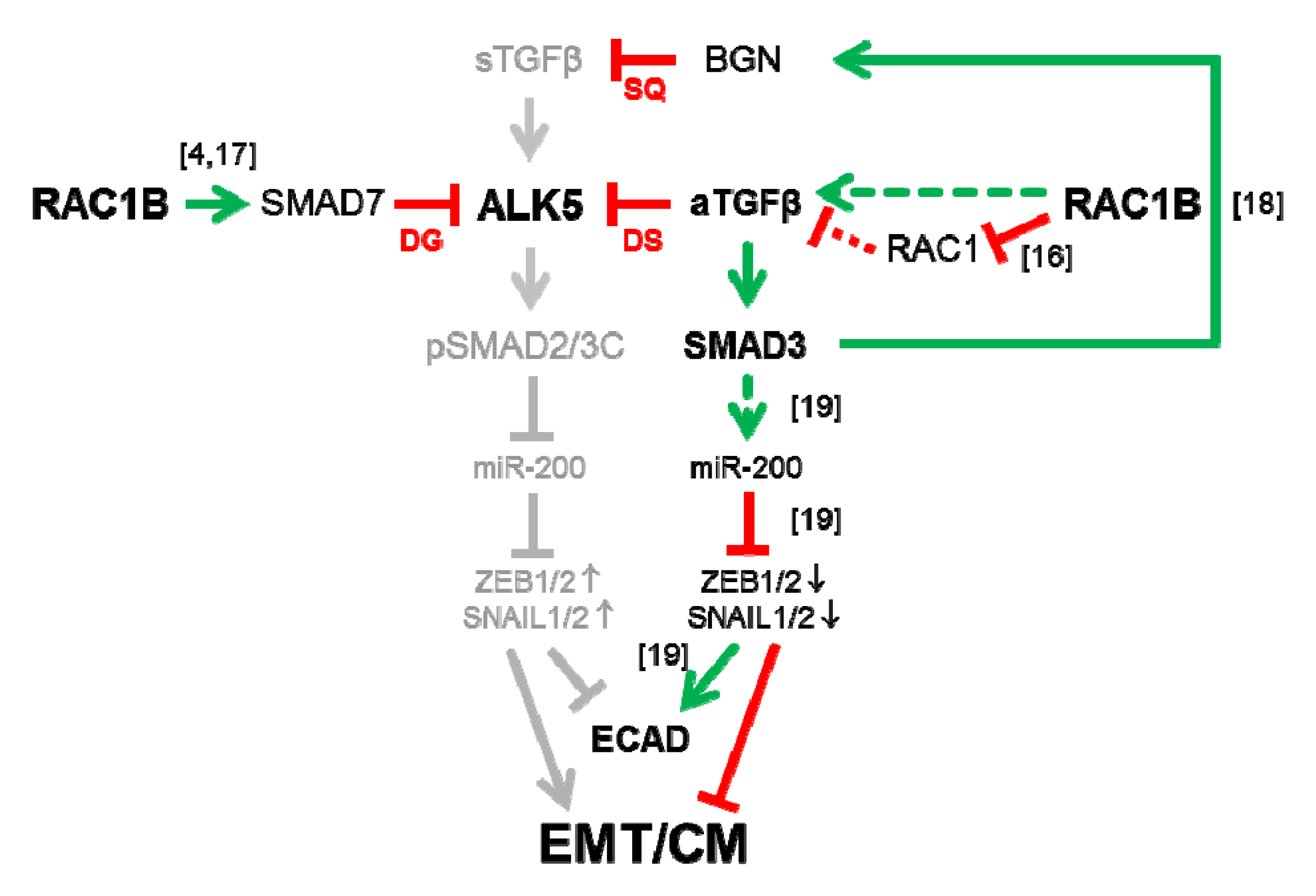
3. Discussion
4. Materials and Methods
4.1. Cells and Generation of Stable Clones Overexpressing RAC1B
4.2. Reagents
4.3. Transient Transfection of siRNA and Expression Vectors
4.4. QPCR Analysis
4.5. Immunoblotting
4.6. Enzyme-Linked Immunosorbant Assay for TGFβ1
4.7. Migration Assays
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellenrieder, V.; Hendler, S.F.; Ruhland, C.; Boeck, W.; Adler, G.; Gress, T.M. TGF-beta-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int. J. Cancer 2001, 93, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Van Obberghen-Schilling, E.; Roche, N.S.; Flanders, K.C.; Sporn, M.B.; Roberts, A.B. Transforming growth factor beta 1 positively regulates its own expression in normal and transformed cells. J. Biol. Chem. 1988, 263, 7741–7746. [Google Scholar] [PubMed]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers 2019, 11, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Carl, C.; Flindt, A.; Hartmann, J.; Dahlke, M.; Rades, D.; Dunst, J.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell. Mol. Life Sci. 2016, 73, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bandyopadhyay, A.; Le, T.; Sun, L. Autocrine TGFbeta supports growth and survival of human breast cancer MDA-MB-231 cells. Oncogene 2002, 21, 7514–7523. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, H.H.; Tang, B.; Wu, A.M.L.; Flanders, K.C.; Moshkovich, N.; Weinberg, D.S.; Welsh, M.A.; Weng, J.; Ochoa, H.J.; et al. The Outcome of TGFβ Antagonism in Metastatic Breast Cancer Models In Vivo Reflects a Complex Balance between Tumor-Suppressive and Proprogression Activities of TGFβ. Clin. Cancer Res. 2020, 26, 643–656. [Google Scholar] [CrossRef] [Green Version]
- Forrester, E.; Chytil, A.; Bierie, B.; Aakre, M.; Gorska, A.E.; Sharif-Afshar, A.R.; Muller, W.J.; Moses, H.L. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005, 65, 2296–2302. [Google Scholar] [CrossRef] [Green Version]
- Warzecha, C.C.; Carstens, R.P. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin. Cancer Biol. 2012, 22, 417–427. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, P.; Rozeboom, B.J.; Aske, J.C.; Dey, N. Active RAC1 Promotes Tumorigenic Phenotypes and Therapy Resistance in Solid Tumors. Cancers 2020, 12, 1541. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, H.L.; Hoffmann, R.; González-López, C.; Doherty, G.J.; Korkola, J.E.; Muñoz-Espín, D. Cellular senescence in cancer: From mechanisms to detection. Mol. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [Green Version]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Kumarasinghe, A.; Musfeldt, M.; Fiedler, C.; Lehnert, H.; Marquardt, J.U. RAC1B Induces SMAD7 via USP26 to Suppress TGFβ1-Dependent Cell Migration in Mesenchymal-Subtype Carcinoma Cells. Cancers 2020, 12, 1545. [Google Scholar] [CrossRef]
- Otterbein, H.; Lehnert, H.; Ungefroren, H. Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan. Cancers 2019, 11, 1959. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.M.; Cha, J.Y.; Kim, J.; Kim, D.; Trang, H.T.; Kim, Y.M.; Cho, Y.H.; Park, D.; Hong, S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene 2012, 31, 3051–3059. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal. 2017, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H. Characterization of autocrine TGFβ expression in pancreatic and mammary tumor cell lines. 2020; manuscript in preparation. [Google Scholar]
- Côme, C.; Magnino, F.; Bibeau, F.; De Santa Barbara, P.; Becker, K.F.; Theillet, C.; Savagner, P. Snail and slug play distinct roles during breast carcinoma progression. Clin. Cancer Res. 2006, 12, 5395–5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.; Kuperwasser, C. SLUG: Critical regulator of epithelial cell identity in breast development and cancer. Cell Adh. Migr. 2014, 8, 578–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fändrich, F.; Röcken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Bakin, A.V.; Arteaga, C.L. Autocrine transforming growth factor-beta signaling mediates Smad-independent motility in human cancer cells. J. Biol. Chem. 2003, 278, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Barnhouse, V.R.; Weist, J.L.; Shukla, V.C.; Ghadiali, S.N.; Kniss, D.A.; Leight, J.L. Myoferlin regulates epithelial cancer cell plasticity and migration through autocrine TGF-β1 signaling. Oncotarget 2018, 9, 19209–19222. [Google Scholar] [CrossRef] [Green Version]
- Christl, J.; Ungefroren, H. RAC1B and RAC1B expression in human mammary epithelial cells. 2020; manuscript in preparation. [Google Scholar]
- Aue, A.; Hinze, C.; Walentin, K.; Ruffert, J.; Yurtdas, Y.; Werth, M.; Chen, W.; Rabien, A.; Kilic, E.; Schulzke, J.D.; et al. A Grainyhead-Like 2/Ovo-Like 2 Pathway Regulates Renal Epithelial Barrier Function and Lumen Expansion. J. Am. Soc. Nephrol. 2015, 26, 2704–2715. [Google Scholar] [CrossRef] [Green Version]
- Nishino, H.; Takano, S.; Yoshitomi, H.; Suzuki, K.; Kagawa, S.; Shimazaki, R.; Shimizu, H.; Furukawa, K.; Miyazaki, M.; Ohtsuka, M. Grainyhead-like 2 (GRHL2) regulates epithelial plasticity in pancreatic cancer progression. Cancer Med. 2017, 6, 2686–2696. [Google Scholar] [CrossRef]
- He, J.; Feng, C.; Zhu, H.; Wu, S.; Jin, P.; Xu, T. Grainyhead-like 2 as a double-edged sword in development and cancer. Am. J. Transl. Res. 2020, 12, 310–331. [Google Scholar]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev. Cell. 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizán, P.; Miller, D.S.; Gori, I.; Das, D.; Schmierer, B.; Hill, C.S. Controlling long-term signaling: Receptor dynamics determine attenuation and refractory behavior of the TGF-β pathway. Sci. Signal. 2013, 6, ra106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ko, C.Y.; Meyers, E.E.; Pedroja, B.S.; Pelaez, N.; Bernstein, A.M. Concentration-dependent effects of transforming growth factor β1 on corneal wound healing. Mol. Vis. 2011, 17, 2835–2846. [Google Scholar]
- Bobr, A.; Igyarto, B.Z.; Haley, K.M.; Li, M.O.; Flavell, R.A.; Kaplan, D.H. Autocrine/paracrine TGF-β1 inhibits Langerhans cell migration. Proc. Natl. Acad. Sci. USA 2012, 109, 10492–10497. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Espinosa, E.; Oemar, B.S.; Lüscher, T.F. Bimodal effects of angiotensin II on migration of human and rat smooth muscle cells. Direct stimulation and indirect inhibition via transforming growth factor-beta 1. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1251–1257. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp. Cell Res. 2005, 305, 292–299. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol. Cancer Res. 2008, 6, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, S.; Mashima, H.; Ohnishi, H.; Sugano, K. IL-13 promotes the proliferation of rat pancreatic stellate cells through the suppression of NF-kappaB/TGF-beta1 pathway. Biochem. Biophys. Res. Commun. 2010, 393, 61–65. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Brabletz, T.; Keck, T. ZEB1 in Pancreatic Cancer. Cancers 2010, 2, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell. 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H. Blockade of TGF-β signaling: A potential target for cancer immunotherapy? Expert Opin. Ther. Targets 2019, 23, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Otterbein, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.-U. RAC1B Regulation of TGFB1 Reveals an Unexpected Role of Autocrine TGFβ1 in the Suppression of Cell Motility. Cancers 2020, 12, 3570. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123570
Ungefroren H, Otterbein H, Wellner UF, Keck T, Lehnert H, Marquardt J-U. RAC1B Regulation of TGFB1 Reveals an Unexpected Role of Autocrine TGFβ1 in the Suppression of Cell Motility. Cancers. 2020; 12(12):3570. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123570
Chicago/Turabian StyleUngefroren, Hendrik, Hannah Otterbein, Ulrich F. Wellner, Tobias Keck, Hendrik Lehnert, and Jens-Uwe Marquardt. 2020. "RAC1B Regulation of TGFB1 Reveals an Unexpected Role of Autocrine TGFβ1 in the Suppression of Cell Motility" Cancers 12, no. 12: 3570. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123570