
Impact of Exercise and Aging on Mitochondrial Homeostasis in Skeletal Muscle: Roles of ROS and Epigenetics


Abstract
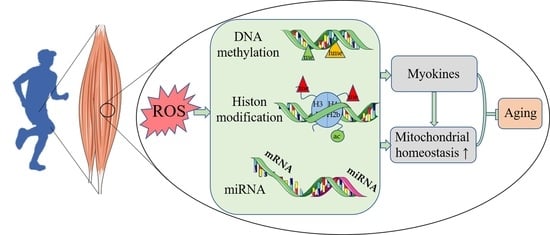
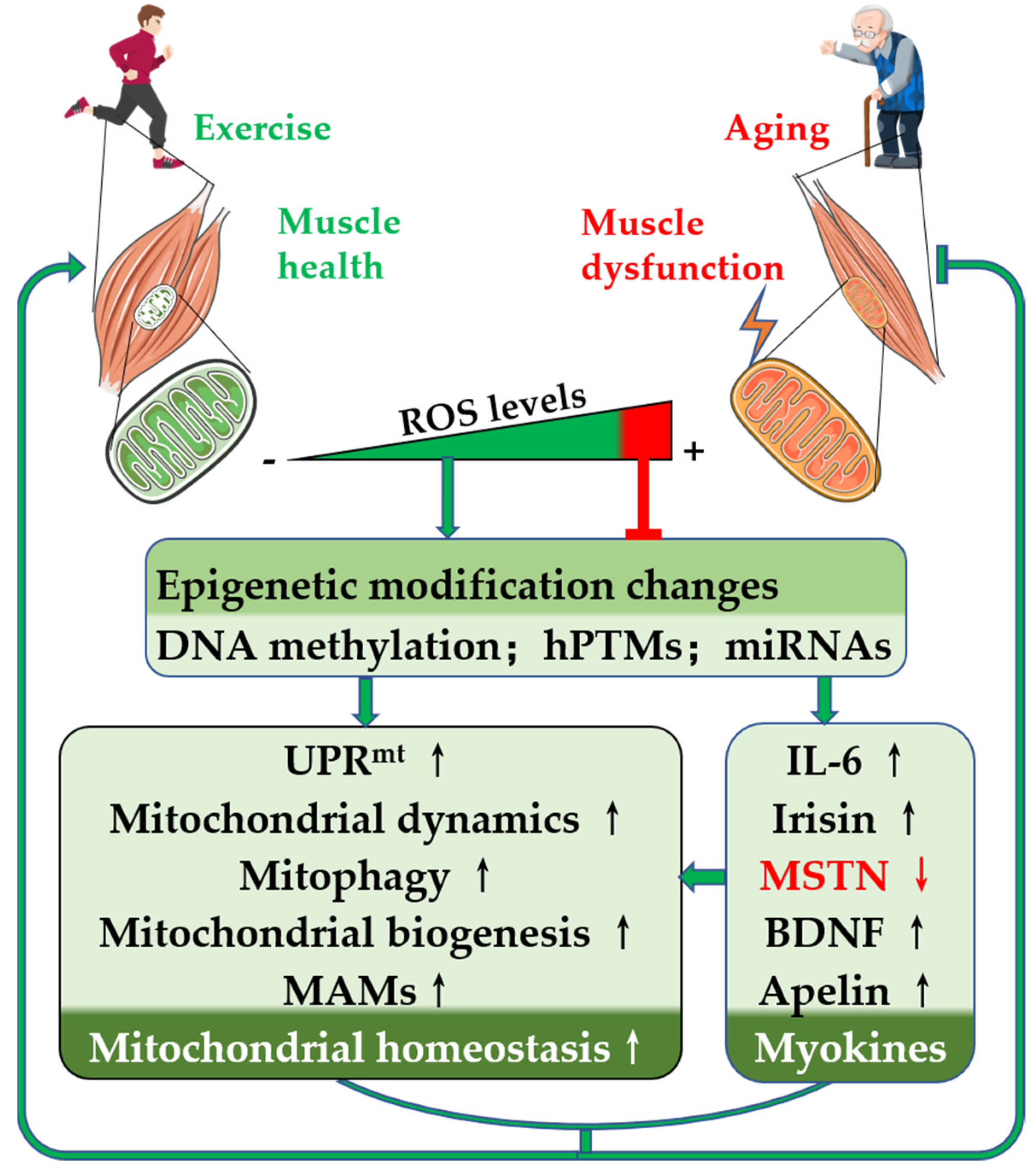
:
{kind=link}
{kind=link}
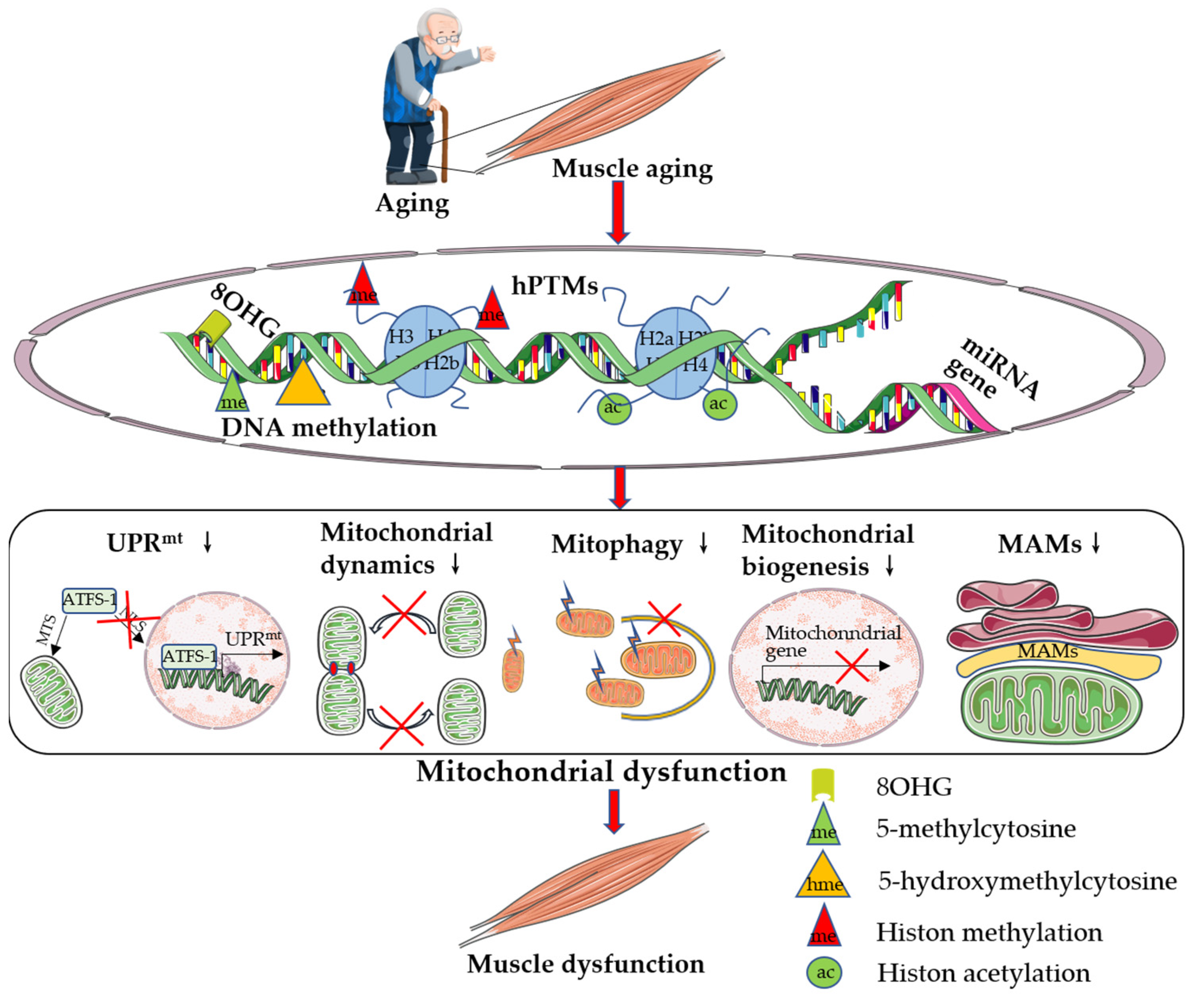
{kind=link}
{kind=link}
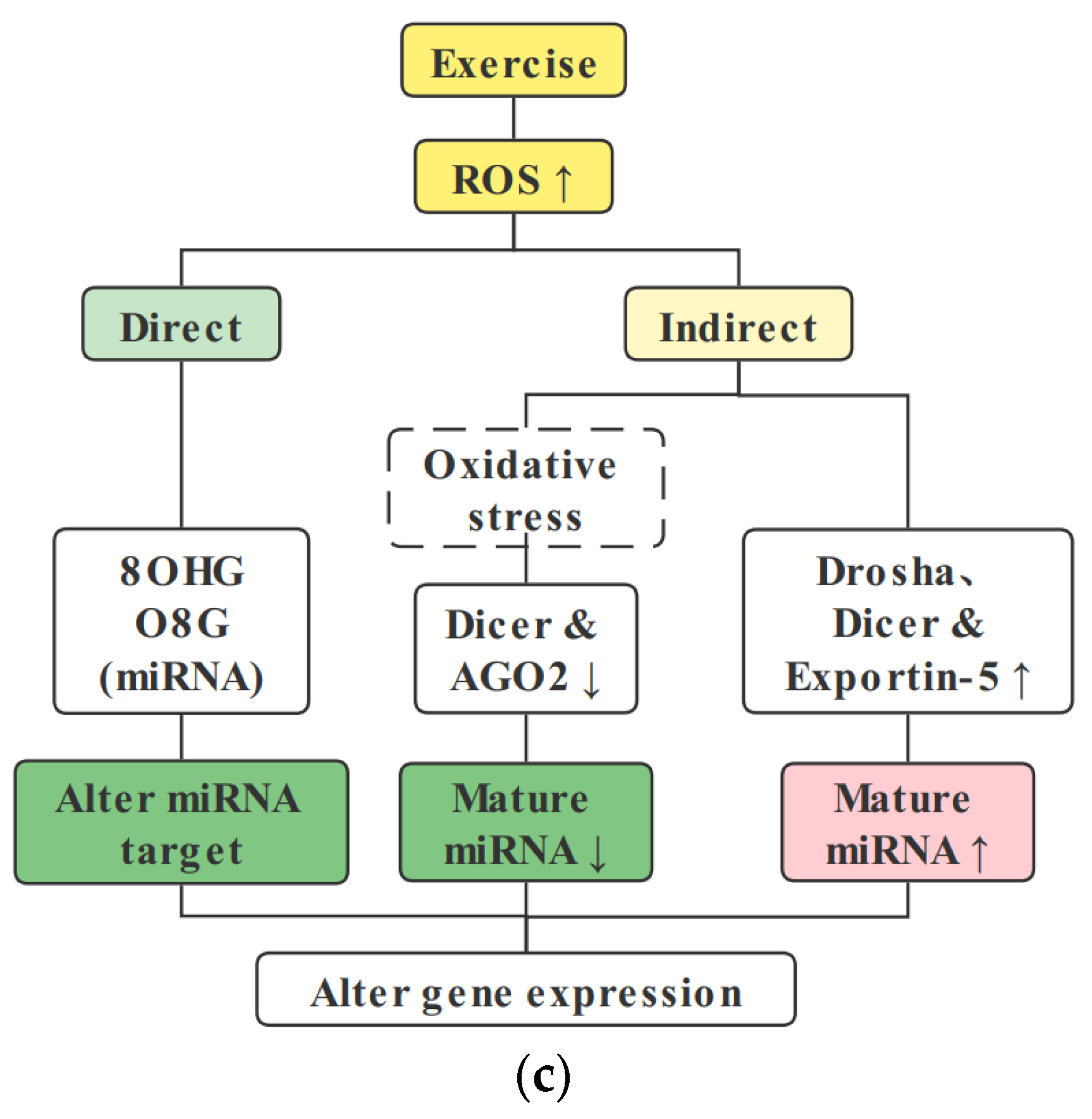
{kind=link}
1. Introduction
2. Aging-Induced Mitochondrial Dysfunction in Skeletal Muscle
2.1. Aging-Associated Reductions in Mitochondrial Biogenesis
2.2. Aging-Associated Alterations in Mitochondrial Dynamics
2.3. Aging-Associated Alterations in Mitophagy
2.4. Aging-Associated Reductions in UPRmt
2.5. Aging- and Exercise-Associated Alterations in MAMs
3. Mitochondria-Associated Epigenetic Changes during Skeletal Muscle Aging
3.1. Aging-Associated Alterations in DNA Methylation
3.2. Aging-Associated Alterations in Histone Posttranslational Modifications
3.3. Aging-Associated Alterations in miRNA Expression
4. Exercise Mitigates Skeletal Muscle Aging via the Regulation of Mitochondria-Associated Epigenetics
4.1. Exercise-Induced Alterations in DNA Methylation
4.2. Exercise-Induced Alterations in Histone Posttranslational Modifications
4.3. Exercise-Induced Alterations in miRNA Expression
5. Role of ROS in the Epigenetic Modification of Skeletal Muscle Mitochondria
5.1. ROS Are Necessary for Exercise-Induced Skeletal Muscle Health Benefits
5.2. ROS Regulate DNA Methylation
5.3. ROS Regulate Histone Posttranslational Modifications
5.4. ROS Regulate miRNA Expression
6. Exercise Modulates Myokine Expression
6.1. Exercise Reverses Myokine Expression during Aging
6.2. Exercise Modulates Myokine Expression via Epigenetic Regulation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Acetyl-CoA | Acetyl-coenzyme A |
| AGO2 | Argonaute-2 |
| BDNF | Brain-derived neurotrophic factor |
| CaMKII | Calmodulin-dependent protein kinase II |
| DNMTs | DNA methyltransferases |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| HDAC | Histone deacetylase |
| HMT | Histone methyltransferase |
| hPTMs | Histone posttranslational modifications |
| IL-6 | Interleukin-6 |
| miRNA | MicroRNA |
| MAMs | Mitochondria-associated endoplasmic reticulum membranes |
| MSTN | Myostatin |
| O8G | 8-oxoguanine |
| 8OHG | 8-oxo-7,8-dihydroguanosine |
| ROS | Reactive oxygen species |
| SAM | S-adenosylmethionine |
| TETs | Ten–eleven translocation enzymes |
| UPRmt | Mitochondrial unfolded protein response |
References
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.; Goodpaster, B.H. Effects of Exercise and Aging on Skeletal Muscle. Cold Spring Harb. Perspect. Med. 2018, 8, a029785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- No, M.-H.; Heo, J.-W.; Yoo, S.-Z.; Kim, C.-J.; Park, D.-H.; Kang, J.-H.; Seo, D.-Y.; Han, J.; Kwak, H.-B. Effects of aging and exercise training on mitochondrial function and apoptosis in the rat heart. Pflugers Arch. 2020, 472, 179–193. [Google Scholar] [CrossRef]
- Goljanek-Whysall, K.; Soriano-Arroquia, A.; McCormick, R.; Chinda, C.; McDonagh, B. miR-181a regulates p62/SQSTM1, parkin, and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle aging. Aging Cell 2020, 19, e13140. [Google Scholar] [CrossRef] [Green Version]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Cartee, G.D.; Hepple, R.T.; Bamman, M.M.; Zierath, J.R. Exercise Promotes Healthy Aging of Skeletal Muscle. Cell Metab. 2016, 23, 1034–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Deng, J.; Qiu, Y.; Gao, J.; Li, J.; Guan, L.; Lee, H.; Zhou, Q.; Xiao, J. Non-coding RNA basis of muscle atrophy. Mol. Ther. Nucleic Acids 2021, 26, 1066–1078. [Google Scholar] [CrossRef]
- Guerville, F.; Barreto, P.D.S.; Ader, I.; Andrieu, S.; Casteilla, L.; Dray, C.; Fazilleau, N.; Guyonnet, S.; Langin, D.; Liblau, R.; et al. Revisiting the hallmarks of aging to identify markers of biological age. J. Prev. Alzheimer’s Dis. 2020, 7, 56–64. [Google Scholar] [CrossRef]
- Carter, H.N.; Pauly, M.; Tryon, L.D.; Hood, D.A. Effect of contractile activity on PGC-1α transcription in young and aged skeletal muscle. J. Appl. Physiol. 2018, 124, 1605–1615. [Google Scholar] [CrossRef]
- Benayoun, B.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, P.; Garcia, P.A.A.; Iyer, C.C.; Popova, L.V.; Arnold, W.D.; Parthun, M.R. Early-onset aging and mitochondrial defects associated with loss of histone acetyltransferase 1 (Hat1). Aging Cell 2019, 18, e12992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Di Vaia, E.; Iaccarino, G. Physical Exercise: A Novel Tool to Protect Mitochondrial Health. Front. Physiol. 2021, 12, 660068. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Hargreaves, M. Exercise adaptations: Molecular mechanisms and potential targets for therapeutic benefit. Nat. Rev. Endocrinol. 2020, 16, 495–505. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, I.; Paronetto, M.P.; Caporossi, D. Exercise, redox homeostasis and the epigenetic landscape. Redox Biol. 2020, 35, 101477. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Jeong, Y.J.; Song, I.-S.; Noh, Y.H.; Seo, K.W.; Kim, M.; Han, J. Glucocorticoid receptor positively regulates transcription of FNDC5 in the liver. Sci. Rep. 2017, 7, 43296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Liu, A.-S.; Zhong, D.; Wang, C.-G.; Yu, M.; Zhang, H.-W.; Xiao, H.; Liu, J.-H.; Zhang, J.; Yin, K. Circular RNA AFF4 modulates osteogenic differentiation in BM-MSCs by activating SMAD1/5 pathway through miR-135a-5p/FNDC5/Irisin axis. Cell Death Dis. 2021, 12, 631. [Google Scholar] [CrossRef]
- Jia, Y.; Gao, G.; Song, H.; Cai, D.; Yang, X.; Zhao, R. Low-protein diet fed to crossbred sows during pregnancy and lactation enhances myostatin gene expression through epigenetic regulation in skeletal muscle of weaning piglets. Eur. J. Nutr. 2016, 55, 1307–1314. [Google Scholar] [CrossRef]
- Elsner, V.R.; Lovatel, G.A.; Moysés, F.; Bertoldi, K.; Spindler, C.; Cechinel, L.R.; Muotri, A.R.; Siqueira, I.R. Exercise induces age-dependent changes on epigenetic parameters in rat hippocampus: A preliminary study. Exp. Gerontol. 2013, 48, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Zhang, Z.; Dong, Z.; Liu, X.; Liu, Y.; Li, X.; Xu, Y.; Guo, Y.; Wang, N.; Zhang, M.; et al. MicroRNA-122-5p Aggravates Angiotensin II-Mediated Myocardial Fibrosis and Dysfunction in Hypertensive Rats by Regulating the Elabela/Apelin-APJ and ACE2-GDF15-Porimin Signaling. J. Cardiovasc. Transl. Res. 2022, 15, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Tsika, R.W.; Herrick, R.E.; Baldwin, K.M. Subunit composition of rodent isomyosins and their distribution in hindlimb skeletal muscles. J. Appl. Physiol. 1987, 63, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Smerdu, V.; Mizrachi, I.K.; Campione, M.; Leinwand, L.; Schiaffino, S. Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am. J. Physiol. Physiol. 1994, 267, C1723–C1728. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Okutsu, M.; Akhtar, Y.N.; Lira, V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011, 110, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Staron, R.S.; Hagerman, F.C.; Hikida, R.S.; Murray, T.F.; Hostler, D.P.; Crill, M.T.; Ragg, K.E.; Toma, K. Fiber Type Composition of the Vastus Lateralis Muscle of Young Men and Women. J. Histochem. Cytochem. 2000, 48, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Bloemberg, D.; Quadrilatero, J. Rapid Determination of Myosin Heavy Chain Expression in Rat, Mouse, and Human Skeletal Muscle Using Multicolor Immunofluorescence Analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef]
- D’Amico, D.; Gammazza, A.M.; Macaluso, F.; Paladino, L.; Scalia, F.; Spinoso, G.; Dimauro, I.; Caporossi, D.; Cappello, F.; Di Felice, V.; et al. Sex-based differences after a single bout of exercise on PGC1α isoforms in skeletal muscle: A pilot study. FASEB J. 2021, 35, e21328. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, R.; Gonzalez, B.; Johnson, S.C. Mitochondrial pathways in human health and aging. Mitochondrion 2020, 54, 72–84. [Google Scholar] [CrossRef]
- Zimmermann, A.; Madreiter-Sokolowski, C.; Stryeck, S.; Abdellatif, M. Targeting the Mitochondria-Proteostasis Axis to Delay Aging. Front. Cell Dev. Biol. 2021, 9, 656201. [Google Scholar] [CrossRef]
- Garcia, S.; Nissanka, N.; Mareco, E.A.; Rossi, S.; Peralta, S.; Díaz, F.; Rotundo, R.L.; Carvalho, R.F.; Moraes, C.T. Overexpression of PGC-1α in aging muscle enhances a subset of young-like molecular patterns. Aging Cell 2018, 17, e12707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Loro, E.; Wada, S.; Kim, B.; Tseng, W.-J.; Li, K.; Khurana, T.S.; Arany, Z. Functional effects of muscle PGC-1alpha in aged animals. Skelet. Muscle 2020, 10, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Faitg, J.; Leduc-Gaudet, J.-P.; Reynaud, O.; Ferland, G.; Gaudreau, P.; Gouspillou, G. Effects of Aging and Caloric Restriction on Fiber Type Composition, Mitochondrial Morphology and Dynamics in Rat Oxidative and Glycolytic Muscles. Front. Physiol. 2019, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Touvier, T.; De Palma, C.; Rigamonti, E.; Scagliola, A.; Incerti, E.; Mazelin, L.; Thomas, J.-L.; D’Antonio, M.; Politi, L.S.; Schaeffer, L.; et al. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 2015, 6, e1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, G.; Romanello, V.; Varanita, T.; Desbats, M.A.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef]
- Beregi, E.; Regius, O.; Hüttl, T.; Göbl, Z. Age-related changes in the skeletal muscle cells. Z. Gerontol. 1988, 21, 83–86. [Google Scholar]
- Beregi, E.; Regius, O. Comparative morphological study of age related mitochondrial changes of the lymphocytes and skeletal muscle cells. Acta Morphol. Hung. 1987, 35, 219–224. [Google Scholar]
- Navratil, M.; Terman, A.; Arriaga, E.A. Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp. Cell Res. 2008, 314, 164–172. [Google Scholar] [CrossRef]
- Taghizadeh, G.; Pourahmad, J.; Mehdizadeh, H.; Foroumadi, A.; Torkaman-Boutorabi, A.; Hassani, S.; Naserzadeh, P.; Shariatmadari, R.; Gholami, M.; Rouini, M.R.; et al. Protective effects of physical exercise on MDMA-induced cognitive and mitochondrial impairment. Free Radic. Biol. Med. 2016, 99, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Flis, D.J.; Olek, R.A.; Kaczor, J.J.; Rodziewicz, E.; Halon, M.; Antosiewicz, J.; Wozniak, M.; Gabbianelli, R.; Ziolkowski, W. Exercise-Induced Changes in Caveolin-1, Depletion of Mitochondrial Cholesterol, and the Inhibition of Mitochondrial Swelling in Rat Skeletal Muscle but Not in the Liver. Oxidative Med. Cell. Longev. 2016, 2016, 3620929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.N.; Richards, B.J.; Slavin, M.; Hood, D.A. Exercise Is Muscle Mitochondrial Medicine. Exerc. Sport Sci. Rev. 2021, 49, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. J. Biol. Inorg. Chem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, A.V.; Peruca, G.F.; Braga, R.R.; Brícola, R.S.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Barbosa, L.T.; et al. High-intensity exercise training induces mitonuclear imbalance and activates the mitochondrial unfolded protein response in the skeletal muscle of aged mice. GeroScience 2021, 43, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.V.; Brícola, R.S.; Braga, R.R.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Simabuco, F.M.; Silva, A.S.R.; et al. Aerobic Exercise Training Induces the Mitonuclear Imbalance and UPRmt in the Skeletal Muscle of Aged Mice. J. Gerontol. Ser. A 2020, 75, 2258–2261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.W.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.B.; Jasper, H. Mitochondrial Proteostasis in the Control of Aging and Longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J.; Li, T.Y. A conserved role of CBP/p300 in mitochondrial stress response and longevity. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Wang, N.; Wang, C.; Zhao, H.; He, Y.; Lan, B.; Sun, L.; Gao, Y. The MAMs Structure and Its Role in Cell Death. Cells 2021, 10, 657. [Google Scholar] [CrossRef]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Sun, C.; Gong, Q.; Feng, D. Mitochondria-Associated Endoplasmic Reticulum Membranes in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 629669. [Google Scholar] [CrossRef] [PubMed]
- Silva-Palacios, A.; Zazueta, C.; Pedraza-Chaverri, J. ER membranes associated with mitochondria: Possible therapeutic targets in heart-associated diseases. Pharmacol. Res. 2020, 156, 104758. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital myopathies: Disorders of excitation–contraction coupling and muscle contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Filippin, L.; Magalhães, P.J.; Di Benedetto, G.; Colella, M.; Pozzan, T. Stable Interactions between Mitochondria and Endoplasmic Reticulum Allow Rapid Accumulation of Calcium in a Subpopulation of Mitochondria. J. Biol. Chem. 2003, 278, 39224–39234. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca2+ Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnoczky, G. Bidirectional Ca2+ dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef] [Green Version]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 2020, 36, 101678. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028.e7. [Google Scholar] [CrossRef] [Green Version]
- Joiner, M.-L.A.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Ashkavand, Z.; Sarasija, S.; Ryan, K.C.; Laboy, J.T.; Norman, K.R. Corrupted ER-mitochondrial calcium homeostasis promotes the collapse of proteostasis. Aging Cell 2020, 19, e13065. [Google Scholar] [CrossRef]
- Cadenas, E.; Boveris, A. Enhancement of hydrogen peroxide formation by protophores and ionophores in antimycin-supplemented mitochondria. Biochem. J. 1980, 188, 31–37. [Google Scholar] [CrossRef]
- Brookes, P.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Gil-Hernández, A.; Silva-Palacios, A. Relevance of endoplasmic reticulum and mitochondria interactions in age-associated diseases. Ageing Res. Rev. 2020, 64, 101193. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria–ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720. [Google Scholar] [CrossRef]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Felipe, A.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated with Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef]
- Merle, A.; Jollet, M.; Britto, F.A.; Goustard, B.; Bendridi, N.; Rieusset, J.; Ollendorff, V.; Favier, F.B. Endurance exercise decreases protein synthesis and ER-mitochondria contacts in mouse skeletal muscle. J. Appl. Physiol. 2019, 127, 1297–1306. [Google Scholar] [CrossRef]
- Flis, D.J.; Dzik, K.; Kaczor, J.J.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Skeletal Muscle Energy Metabolism, Oxidative Stress, and Mitochondrial Cholesterol Content in Amyotrophic Lateral Sclerosis Mice. Oxidative Med. Cell. Longev. 2018, 2018, 5940748. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.J.; Place, N.; Westerblad, H. Molecular Basis for Exercise-Induced Fatigue: The Importance of Strictly Controlled Cellular Ca2+ Handling. Cold Spring Harb. Perspect. Med. 2018, 8, a029710. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca2+ transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Talens, R.P.; Christensen, K.; Putter, H.; Willemsen, G.; Christiansen, L.; Kremer, D.; Suchiman, H.E.D.; Slagboom, P.; Boomsma, D.I.; Heijmans, B.T. Epigenetic variation during the adult lifespan: Cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell 2012, 11, 694–703. [Google Scholar] [CrossRef]
- Turner, D.C.; Gorski, P.P.; Maasar, M.F.; Seaborne, R.A.; Baumert, P.; Brown, A.D.; Kitchen, M.O.; Erskine, R.M.; Dos-Remedios, I.; Voisin, S.; et al. DNA methylation across the genome in aged human skeletal muscle tissue and muscle-derived cells: The role of HOX genes and physical activity. Sci. Rep. 2020, 10, 15360. [Google Scholar] [CrossRef]
- Sailani, M.R.; Halling, J.F.; Møller, H.D.; Lee, H.; Plomgaard, P.; Pilegaard, H.; Snyder, M.P.; Regenberg, B. Lifelong physical activity is associated with promoter hypomethylation of genes involved in metabolism, myogenesis, contractile properties and oxidative stress resistance in aged human skeletal muscle. Sci. Rep. 2019, 9, 3272. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Davegårdh, C.; Wedin, E.H.; Broholm, C.; Henriksen, T.; Pedersen, M.; Pedersen, B.K.; Scheele, C.; Ling, C. Sex influences DNA methylation and gene expression in human skeletal muscle myoblasts and myotubes. Stem Cell Res. Ther. 2019, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Koczor, C.A.; Ludlow, I.; Fields, E.J.; Jiao, Z.; Ludaway, T.; Russ, R.; Lewis, W. Mitochondrial polymerase gamma dysfunction and aging cause cardiac nuclear DNA methylation changes. Physiol. Genom. 2016, 48, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Hua, X.-M.; Wang, J.; Qian, D.-M.; Song, J.-Y.; Chen, H.; Zhu, X.-L.; Zhou, R.; Zhao, Y.-D.; Zhou, X.-Z.; Li, L.; et al. DNA methylation level of promoter region of activating transcription factor 5 in glioma. J. Zhejiang Univ. Sci. B 2015, 16, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Xia, Y.; Wang, J.; Lin, Z.; Ou, Y.; Liu, X.; Liu, W.; Zhou, B.; Luo, H.; Zhou, B.; et al. Integrated analyses of DNA methylation and hydroxymethylation reveal tumor suppressive roles of ECM1, ATF5, and EOMESin human hepatocellular carcinoma. Genome Biol. 2014, 15, 533. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPRmt. Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [Green Version]
- Li, T.Y.; Sleiman, M.B.; Li, H.; Gao, A.W.; Mottis, A.; Bachmann, A.M.; El Alam, G.; Li, X.; Goeminne, L.J.E.; Schoonjans, K.; et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 2021, 1, 165–178. [Google Scholar] [CrossRef]
- Shao, L.-W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.-Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef]
- Drummond, M.J.; McCarthy, J.J.; Fry, C.S.; Esser, K.A.; Rasmussen, B.B. Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am. J. Physiol. Metab. 2008, 295, E1333–E1340. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Liu, Y.; Li, Q.; Zhang, J.; Ge, C.; Wang, G.; Chen, G.; Liu, D.; Yang, F. Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age. Genes 2020, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Enielsen, S.; Ehvid, T.; Ekelly, M.; Elindegaard, B.; Edethlefsen, C.; Ewinding, K.; Emathur, N.; Escheele, C.; Pedersen, B.K.; Laye, M.J. Muscle specific miRNAs are induced by testosterone and independently upregulated by age. Front. Physiol. 2013, 4, 394. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; Sato, Y.; Wang, C.; Yue, F.; Kuang, S.; Gavin, T.P. Impaired exercise tolerance, mitochondrial biogenesis, and muscle fiber maintenance in miR-133a–deficient mice. FASEB J. 2016, 30, 3745–3758. [Google Scholar] [CrossRef] [Green Version]
- Dahlmans, D.; Houzelle, A.; Andreux, P.; Wang, X.; Jörgensen, J.A.; Moullan, N.; Daemen, S.; Kersten, S.; Auwerx, J.; Hoeks, J. MicroRNA-382 silencing induces a mitonuclear protein imbalance and activates the mitochondrial unfolded protein response in muscle cells. J. Cell. Physiol. 2019, 234, 6601–6610. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.; Wang, X.; Matarese, A.; Chavez, C.; Santulli, G. Dual Microrna-Targeting Rescues the Impaired Mitochondrial Unfolded Protein Response in Heart Failure. Circulation 2019, 140, A16384. [Google Scholar]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute Exercise Remodels Promoter Methylation in Human Skeletal Muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Bajpeyi, S.; Covington, J.D.; Taylor, E.M.; Stewart, L.K.; Galgani, J.E.; Henagan, T.M. Skeletal Muscle PGC1α −1 Nucleosome Position and −260 nt DNA Methylation Determine Exercise Response and Prevent Ectopic Lipid Accumulation in Men. Endocrinology 2017, 158, 2190–2199. [Google Scholar] [CrossRef]
- Hunter, D.J.; James, L.; Hussey, B.; Wadley, A.J.; Lindley, M.R.; Mastana, S.S. Impact of aerobic exercise and fatty acid supplementation on global and gene-specific DNA methylation. Epigenetics 2019, 14, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Landen, S.; Voisin, S.; Craig, J.M.; McGee, S.L.; Lamon, S.; Eynon, N. Genetic and epigenetic sex-specific adaptations to endurance exercise. Epigenetics 2019, 14, 523–535. [Google Scholar] [CrossRef]
- Brown, W. Exercise-associated DNA methylation change in skeletal muscle and the importance of imprinted genes: A bioinformatics meta-analysis. Br. J. Sports Med. 2015, 49, 1567–1578. [Google Scholar] [CrossRef] [Green Version]
- Maasar, M.-F.; Turner, D.C.; Gorski, P.P.; Seaborne, R.A.; Strauss, J.A.; Shepherd, S.O.; Cocks, M.; Pillon, N.J.; Zierath, J.R.; Hulton, A.T.; et al. The Comparative Methylome and Transcriptome After Change of Direction Compared to Straight Line Running Exercise in Human Skeletal Muscle. Front. Physiol. 2021, 12, 619447. [Google Scholar] [CrossRef]
- Rasmussen, M.; Zierath, J.; Barrès, R. Dynamic epigenetic responses to muscle contraction. Drug Discov. Today 2014, 19, 1010–1014. [Google Scholar] [CrossRef]
- Small, L.; Ingerslev, L.R.; Manitta, E.; Laker, R.C.; Hansen, A.N.; Deeney, B.; Carrié, A.; Couvert, P.; Barrès, R. Ablation of DNA-methyltransferase 3A in skeletal muscle does not affect energy metabolism or exercise capacity. PLoS Genet. 2021, 17, e1009325. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Thomas, R.R.; Bennett, J.P.; Taylor, S.M. Epigenetic Modifications of the PGC-1α Promoter during Exercise Induced Expression in Mice. PLoS ONE 2015, 10, e0129647. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.S.; Anand, K.; Malindisa, S.T.; Oladipo, A.O.; Fagbohun, O.F. Exercise, CaMKII, and type 2 diabetes. EXCLI J. 2021, 20, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.S.; Ayeleso, A.O.; Mukwevho, E. Exercise increases hyper-acetylation of histones on the Cis -element of NRF-1 binding to the Mef2a promoter: Implications on type 2 diabetes. Biochem. Biophys. Res. Commun. 2017, 486, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Czubryt, M.P.; McAnally, J.; Fishman, G.I.; Olson, E.N. Regulation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2003, 100, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Masuzawa, R.; Konno, R.; Ohsawa, I.; Watanabe, A.; Kawano, F. Muscle type-specific RNA polymerase II recruitment during PGC-1α gene transcription after acute exercise in adult rats. J. Appl. Physiol. 2018, 125, 1238–1245. [Google Scholar] [CrossRef]
- Jacques, M.; Hiam, D.; Craig, J.; Barrès, R.; Eynon, N.; Voisin, S. Epigenetic changes in healthy human skeletal muscle following exercise—A systematic review. Epigenetics 2019, 14, 633–648. [Google Scholar] [CrossRef]
- Russell, A.P.; Lamon, S.; Boon, H.; Wada, S.; Güller, I.; Brown, E.L.; Chibalin, A.V.; Zierath, J.R.; Snow, R.J.; Stepto, N.I.; et al. Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J. Physiol. 2013, 591, 4637–4653. [Google Scholar] [CrossRef]
- Rodrigues, A.C.; Spagnol, A.R.; Frias, F.D.T.; de Mendonça, M.; Araújo, H.N.; Guimarães, D.; Silva, W.J.; Bolin, A.P.; Murata, G.M.; Silveira, L. Intramuscular Injection of miR-1 Reduces Insulin Resistance in Obese Mice. Front. Physiol. 2021, 12, 676265. [Google Scholar] [CrossRef]
- Sun, Y.; Cui, D.; Zhang, Z.; Zhang, Q.; Ji, L.; Ding, S. Voluntary wheel exercise alters the levels of miR-494 and miR-696 in the skeletal muscle of C57BL/6 mice. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2016, 202, 16–22. [Google Scholar] [CrossRef]
- Massart, J.; Sjögren, R.J.O.; Egan, B.; Garde, C.; Lindgren, M.; Gu, W.; Ferreira, D.M.S.; Katayama, M.; Ruas, J.L.; Barrès, R.; et al. Endurance exercise training-responsive miR-19b-3p improves skeletal muscle glucose metabolism. Nat. Commun. 2021, 12, 5948. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Jacob, P.; Middleton, F.A.; Pérez, O.; Gagnon, Z. Distance running alters peripheral microRNAs implicated in metabolism, fluid balance, and myosin regulation in a sex-specific manner. Physiol. Genom. 2018, 50, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, N.; Parbin, S.; Kar, S.; Das, L.; Kirtana, R.; Seshadri, G.S.; Sengupta, D.; Deb, M.; Kausar, C.; Patra, S.K. Epigenetic silencing of genes enhanced by collective role of reactive oxygen species and MAPK signaling downstream ERK/Snail axis: Ectopic application of hydrogen peroxide repress CDH1 gene by enhanced DNA methyltransferase activity in human breast cancer. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1651–1665. [Google Scholar] [CrossRef]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Rosini, E.; Pollegioni, L. Reactive oxygen species as a double-edged sword: The role of oxidative enzymes in antitumor therapy. BioFactors 2022, 48, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.Y.; Miralda, I.; Armstrong, C.L.; Uriarte, S.M.; Bagaitkar, J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol. Oral Microbiol. 2019, 34, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Taherkhani, S.; Suzuki, K.; Castell, L. A Short Overview of Changes in Inflammatory Cytokines and Oxidative Stress in Response to Physical Activity and Antioxidant Supplementation. Antioxidants 2020, 9, 886. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Bo, H.; Song, Y.; Li, C.; Zhang, Y. Mitochondrial ROS Produced by Skeletal Muscle Mitochondria Promote the Decisive Signal for UPRmt Activation. BioMed Res. Int. 2022, 2022, 7436577. [Google Scholar] [CrossRef]
- Thirupathi, A.; Pinho, R.A.; Chang, Y.-Z. Physical exercise: An inducer of positive oxidative stress in skeletal muscle aging. Life Sci. 2020, 252, 117630. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.I.; Tarnopolsky, M.A. Mitochondria and Aging—The Role of Exercise as a Countermeasure. Biology 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, M.; Gaetani, S.; Monaco, F.; Neuzil, J.; Santarelli, L. Epigenetic Regulation of miRNA Expression in Malignant Mesothelioma: miRNAs as Biomarkers of Early Diagnosis and Therapy. Front. Oncol. 2019, 9, 1293. [Google Scholar] [CrossRef]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Le, D.D.; Fujimori, D.G. Protein and nucleic acid methylating enzymes: Mechanisms and regulation. Curr. Opin. Chem. Biol. 2012, 16, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulter, J.B.; O’Driscoll, C.M.; Bressler, J.P. Hydroquinone Increases 5-Hydroxymethylcytosine Formation through Ten Eleven Translocation 1 (TET1) 5-Methylcytosine Dioxygenase. J. Biol. Chem. 2013, 288, 28792–28800. [Google Scholar] [CrossRef] [Green Version]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Thirupathi, A.; De Souza, C.T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Stephens, N.A.; Brouwers, B.; Eroshkin, A.M.; Yi, F.; Cornnell, H.H.; Meyer, C.; Goodpaster, B.H.; Pratley, R.E.; Smith, S.R.; Sparks, L.M. Exercise Response Variations in Skeletal Muscle PCr Recovery Rate and Insulin Sensitivity Relate to Muscle Epigenomic Profiles in Individuals With Type 2 Diabetes. Diabetes Care 2018, 41, 2245–2254. [Google Scholar] [CrossRef] [Green Version]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.-S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef] [Green Version]
- Marmisolle, I.; Martínez, J.; Liu, J.; Mastrogiovanni, M.; Fergusson, M.M.; Rovira, I.I.; Castro, L.; Trostchansky, A.; Moreno, M.; Cao, L.; et al. Reciprocal regulation of acetyl-CoA carboxylase 1 and senescence in human fibroblasts involves oxidant mediated p38 MAPK activation. Arch. Biochem. Biophys. 2017, 613, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Morales-Alamo, D.; Guerra, B.; Ponce-González, J.G.; Guadalupe-Grau, A.; Santana, A.; Martin-Rincon, M.; Gelabert-Rebato, M.; Cadefau, J.A.; Cusso, R.; Dorado, C.; et al. Skeletal muscle signaling, metabolism, and performance during sprint exercise in severe acute hypoxia after the ingestion of antioxidants. J. Appl. Physiol. 2017, 123, 1235–1245. [Google Scholar] [CrossRef]
- Miotto, P.M.; Holloway, G.P. Exercise-induced reductions in mitochondrial ADP sensitivity contribute to the induction of gene expression and mitochondrial biogenesis through enhanced mitochondrial H2O2 emission. Mitochondrion 2019, 46, 116–122. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Epigenetics and Exercise. Trends Endocrinol. Metab. 2019, 30, 636–645. [Google Scholar] [CrossRef]
- Elliott, P.J.; Jirousek, M. Sirtuins: Novel targets for metabolic disease. Curr. Opin. Investig. Drugs 2008, 9, 371–378. [Google Scholar]
- Yang, Y.; Fu, W.; Chen, J.; Olashaw, N.; Zhang, X.; Nicosia, S.V.; Bhalla, K.; Bai, W. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol. 2007, 9, 1253–1262. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [CrossRef]
- Wang, J.-X.; Gao, J.; Ding, S.-L.; Wang, K.; Jiao, J.-Q.; Wang, Y.; Sun, T.; Zhou, L.-Y.; Long, B.; Zhang, X.-J.; et al. Oxidative Modification of miR-184 Enables It to Target Bcl-xL and Bcl-w. Mol. Cell 2015, 59, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Seok, H.; Lee, H.; Lee, S.; Ahn, S.H.; Lee, H.-S.; Kim, G.-W.D.; Peak, J.; Park, J.; Cho, Y.K.; Jeong, Y.; et al. Position-specific oxidation of miR-1 encodes cardiac hypertrophy. Nature 2020, 584, 279–285. [Google Scholar] [CrossRef]
- Natarelli, L.; Weber, C. A Non-Canonical Link between Non-Coding RNAs and Cardiovascular Diseases. Biomedicines 2022, 10, 445. [Google Scholar] [CrossRef]
- Emde, A.; Hornstein, E. mi RNA s at the interface of cellular stress and disease. EMBO J. 2014, 33, 1428–1437. [Google Scholar] [CrossRef] [Green Version]
- Druz, A.; Betenbaugh, M.; Shiloach, J. Glucose depletion activates mmu-miR-466h-5p expression through oxidative stress and inhibition of histone deacetylation. Nucleic Acids Res. 2012, 40, 7291–7302. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.-R.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Safdar, A.; Abadi, A.; Akhtar, M.; Hettinga, B.P.; Tarnopolsky, M.A. miRNA in the Regulation of Skeletal Muscle Adaptation to Acute Endurance Exercise in C57Bl/6J Male Mice. PLoS ONE 2009, 4, e5610. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Fischer, C. Beneficial health effects of exercise—The role of IL-6 as a myokine. Trends Pharmacol. Sci. 2007, 28, 152–156. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscle as a Secretory Organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Chow, L.S.; Gerszten, R.E.; Taylor, J.M.; Pedersen, B.K.; van Praag, H.; Trappe, S.; Febbraio, M.A.; Galis, Z.S.; Gao, Y.; Haus, J.M.; et al. Exerkines in health, resilience and disease. Nat. Rev. Endocrinol. 2022, 18, 273–289. [Google Scholar] [CrossRef]
- Bo, H.; Jiang, N.; Zhang, Z.-Y.; Ji, L.-L.; Zhang, Y. Exercise and health: From evaluation of health-promoting effects of exercise to exploration of exercise mimetics. Sheng Li Ke Xue Jin Zhan [Prog. Physiol.] 2014, 45, 251–256. [Google Scholar]
- Exercise Metabolism. Cell Metab. 2015, 22, 18–24. [CrossRef] [Green Version]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [Green Version]
- Mangano, G.D.; Fouani, M.; D’Amico, D.; Di Felice, V.; Barone, R. Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training. Int. J. Mol. Sci. 2022, 23, 3004. [Google Scholar] [CrossRef]
- Nara, H.; Watanabe, R. Anti-Inflammatory Effect of Muscle-Derived Interleukin-6 and Its Involvement in Lipid Metabolism. Int. J. Mol. Sci. 2021, 22, 9889. [Google Scholar] [CrossRef]
- Daou, H.N. Exercise as an anti-inflammatory therapy for cancer cachexia: A focus on interleukin-6 regulation. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R296–R310. [Google Scholar] [CrossRef]
- Pedersen, B.K. Anti-inflammatory effects of exercise: Role in diabetes and cardiovascular disease. Eur. J. Clin. Investig. 2017, 47, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Padilha, C.S.; Borges, F.H.; Da Silva, L.E.C.M.; Frajacomo, F.T.T.; Jordao, A.A.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; Deminice, R. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro-inflammatory state, and cachexia in Walker-256 tumor-bearing rats. Appl. Physiol. Nutr. Metab. 2017, 42, 916–923. [Google Scholar] [CrossRef]
- Santos, J.D.M.B.D.; Bachi, A.L.L.; Junior, L.A.L.; Foster, R.; Sierra, A.P.R.; Benetti, M.; Araújo, J.R.; Ghorayeb, N.; Kiss, M.A.P.D.; Vieira, R.P.; et al. The Relationship of IL-8 and IL-10 Myokines and Performance in Male Marathon Runners Presenting Exercise-Induced Bronchoconstriction. Int. J. Environ. Res. Public Health 2020, 17, 2622. [Google Scholar] [CrossRef]
- Li, L.; Mühlfeld, C.; Niemann, B.; Pan, R.; Li, R.; Hilfiker-Kleiner, D.; Chen, Y.; Rohrbach, S. Mitochondrial biogenesis and PGC-1α deacetylation by chronic treadmill exercise: Differential response in cardiac and skeletal muscle. Basic Res. Cardiol. 2011, 106, 1221–1234. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Zhang, R.; Fu, T.; Zhao, X.; Qiu, Y.; Hu, X.; Shi, H.; Yin, X. Association of Circulating Irisin Levels with Adiposity and Glucose Metabolic Profiles in a Middle-Aged Chinese Population: A Cross-Sectional Study. Diabetes Metab. Syndr. Obesity Targets Ther. 2020, 13, 4105–4112. [Google Scholar] [CrossRef]
- Planella-Farrugia, C.; Comas, F.; Sabater-Masdeu, M.; Moreno, M.; Moreno-Navarrete, J.M.; Rovira, O.; Ricart, W.; Fernández-Real, J.M. Circulating Irisin and Myostatin as Markers of Muscle Strength and Physical Condition in Elderly Subjects. Front. Physiol. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto-Mikami, E.; Sato, K.; Kurihara, T.; Hasegawa, N.; Fujie, S.; Fujita, S.; Sanada, K.; Hamaoka, T.; Tabata, I.; Iemitsu, M. Endurance Training-Induced Increase in Circulating Irisin Levels Is Associated with Reduction of Abdominal Visceral Fat in Middle-Aged and Older Adults. PLoS ONE 2015, 10, e0120354. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; So, B.; Choi, M.; Kang, D.; Song, W. Resistance exercise training increases the expression of irisin concomitant with improvement of muscle function in aging mice and humans. Exp. Gerontol. 2015, 70, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Amanat, S.; Sinaei, E.; Panji, M.; MohammadporHodki, R.; Bagheri-Hosseinabadi, Z.; Asadimehr, H.; Fararouei, M.; Dianatinasab, A. A Randomized Controlled Trial on the Effects of 12 Weeks of Aerobic, Resistance, and Combined Exercises Training on the Serum Levels of Nesfatin-1, Irisin-1 and HOMA-IR. Front. Physiol. 2020, 11, 562895. [Google Scholar] [CrossRef]
- Belviranlı, M.; Okudan, N. Exercise training increases cardiac, hepatic and circulating levels of brain-derived neurotrophic factor and irisin in young and aged rats. Horm. Mol. Biol. Clin. Investig. 2018, 36, 3. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Ando, D.; Takamatsu, K.; Goto, K. Resistance exercise induces a greater irisin response than endurance exercise. Metabolism 2015, 64, 1042–1050. [Google Scholar] [CrossRef]
- He, W.; Wang, P.; Chen, Q.; Li, C. Exercise enhances mitochondrial fission and mitophagy to improve myopathy following critical limb ischemia in elderly mice via the PGC1a/FNDC5/irisin pathway. Skelet. Muscle 2020, 10, 25. [Google Scholar] [CrossRef]
- Gomes, M.J.; Martinez, P.F.; Pagan, L.U.; Damatto, R.L.; Cezar, M.D.M.; Lima, A.R.R.; Okoshi, K.; Okoshi, M.P. Skeletal muscle aging: Influence of oxidative stress and physical exercise. Oncotarget 2017, 8, 20428–20440. [Google Scholar] [CrossRef] [Green Version]
- McPherron, A.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Shabkhiz, F.; Khalafi, M.; Rosenkranz, S.; Karimi, P.; Moghadami, K. Resistance training attenuates circulating FGF-21 and myostatin and improves insulin resistance in elderly men with and without type 2 diabetes mellitus: A randomised controlled clinical trial. Eur. J. Sport Sci. 2021, 21, 636–645. [Google Scholar] [CrossRef]
- Ryan, A.; Li, G.; Blumenthal, J.; Ortmeyer, H. Aerobic exercise + weight loss decreases skeletal muscle myostatin expression and improves insulin sensitivity in older adults. Obesity 2013, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Jerobin, J.; Ramanjaneya, M.; Bettahi, I.; Parammal, R.; Siveen, K.S.; Alkasem, M.; Aye, M.; Sathyapalan, T.; Skarulis, M.; Atkin, S.L.; et al. Regulation of circulating CTRP-2/CTRP-9 and GDF-8/GDF-15 by intralipids and insulin in healthy control and polycystic ovary syndrome women following chronic exercise training. Lipids Health Dis. 2021, 20, 34. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Zhao, M.; Li, J.; Li, S.; Zhu, S.; Yao, X.; Gao, X.; Yang, S. Myostatin is involved in skeletal muscle dysfunction in chronic obstructive pulmonary disease via Drp-1 mediated abnormal mitochondrial division. Ann. Transl. Med. 2022, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.; Cannavo, A.; Gambino, G.; Cimini, M.; Ferrara, N.; Kishore, R.; Paolocci, N.; Rengo, G. Aging is associated with cardiac autonomic nerve fiber depletion and reduced cardiac and circulating BDNF levels. J. Geriatr. Cardiol. 2021, 18, 549–559. [Google Scholar] [CrossRef]
- Belviranlı, M.; Okudan, N. Exercise Training Protects Against Aging-Induced Cognitive Dysfunction via Activation of the Hippocampal PGC-1α/FNDC5/BDNF Pathway. NeuroMolecular Med. 2018, 20, 386–400. [Google Scholar] [CrossRef]
- Matthews, V.B.; Åström, M.-B.; Chan, S.; Bruce, C.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Brobst, D.; Chan, W.S.; Tse, M.C.L.; Herlea-Pana, O.; Ahuja, P.; Bi, X.; Zaw, A.M.; Kwong, Z.S.W.; Jia, W.-H.; et al. Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal. 2019, 12, eaau1468. [Google Scholar] [CrossRef]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradère, J.-P.; Le Gonidec, S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef]
- Dundar, A.; Kocahan, S.; Sahin, L. Associations of apelin, leptin, irisin, ghrelin, insulin, glucose levels, and lipid parameters with physical activity during eight weeks of regular exercise training. Arch. Physiol. Biochem. 2021, 127, 291–295. [Google Scholar] [CrossRef]
- Son, J.S.; Chae, S.A.; Wang, H.; Chen, Y.; Iniguez, A.B.; de Avila, J.M.; Jiang, Z.; Zhu, M.-J.; Du, M. Maternal Inactivity Programs Skeletal Muscle Dysfunction in Offspring Mice by Attenuating Apelin Signaling and Mitochondrial Biogenesis. Cell Rep. 2020, 33, 108461. [Google Scholar] [CrossRef]
- Coco, M.; Perciavalle, V.; Cavallari, P.; Bolzoni, F.; Graziano, A.C.E.; Perciavalle, V. Effects of age and sex on epigenetic modification induced by an acute physical exercise. Medicine 2017, 96, e8325. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, O.; Brecklinghaus, T.; Dille, M.; Springer, C.; de Wendt, C.; Altenhofen, D.; Binsch, C.; Knebel, B.; Scheller, J.; Hardt, C.; et al. Histone deacetylase 5 regulates interleukin 6 secretion and insulin action in skeletal muscle. Mol. Metab. 2020, 42, 101062. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, P.; Wei, W.; Wang, C.; Zhong, Y.; Lv, L.; Wang, J. Serum MicroRNA Expression Patterns in Subjects after the 5-km Exercise Are Strongly Associated with Cardiovascular Adaptation. Front. Physiol. 2021, 12, 755656. [Google Scholar] [CrossRef]
- Dorhoi, A.; Iannaccone, M.; Farinacci, M.; Faé, K.C.; Schreiber, J.; Moura-Alves, P.; Nouailles, G.; Mollenkopf, H.-J.; Oberbeck-Müller, D.; Jörg, S.; et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J. Clin. Investig. 2013, 123, 4836–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; He, Y.; Zhou, Z.; Ramirez, T.; Gao, Y.; Gao, Y.; Ross, R.A.; Cao, H.; Cai, Y.; Xu, M.; et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6–p47phox–oxidative stress pathway in neutrophils. Gut 2017, 66, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Tong, Y.; Wu, N.; Wan, G.-W.; Zheng, F.; Chen, J.-Y.; Lei, J.-Z.; Zhou, H.; Chen, A.-D.; Wang, J.-J.; et al. Inhibition of miR-135a-5p attenuates vascular smooth muscle cell proliferation and vascular remodeling in hypertensive rats. Acta Pharmacol. Sin. 2021, 42, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Espinal, M.P.; Gasperini, C.; Marzi, M.; Braccia, C.; Armirotti, A.; Pötzsch, A.; Walker, T.L.; Fabel, K.; Nicassio, F.; Kempermann, G.; et al. MiR-135a-5p Is Critical for Exercise-Induced Adult Neurogenesis. Stem Cell Rep. 2019, 12, 1298–1312. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Zhang, R.; Tesfaye, D.; Tholen, E.; Looft, C.; Hölker, M.; Schellander, K.; Cinar, M.U. Sulforaphane causes a major epigenetic repression of myostatin in porcine satellite cells. Epigenetics 2012, 7, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; EL Andaloussi, S.; Morris, K.V.; McClorey, G.; Wood, M.J. Small RNA-Mediated Epigenetic Myostatin Silencing. Mol. Ther. Nucleic Acids 2012, 1, e23. [Google Scholar] [CrossRef]
- Zarfeshani, A.; Ngo, S.; Sheppard, A.M. Leucine alters hepatic glucose/lipid homeostasis via the myostatin-AMP-activated protein kinase pathway—Potential implications for nonalcoholic fatty liver disease. Clin. Epigenetics 2014, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Drummond, M.J.; Glynn, E.L.; Fry, C.S.; Dhanani, S.; Volpi, E.; Rasmussen, B.B. Essential Amino Acids Increase MicroRNA-499, -208b, and -23a and Downregulate Myostatin and Myocyte Enhancer Factor 2C mRNA Expression in Human Skeletal Muscle. J. Nutr. 2009, 139, 2279–2284. [Google Scholar] [CrossRef]
- Tomiga, Y.; Sakai, K.; Ra, S.; Kusano, M.; Ito, A.; Uehara, Y.; Takahashi, H.; Kawanaka, K.; Soejima, H.; Higaki, Y. Short-term running exercise alters DNA methylation patterns in neuronal nitric oxide synthase and brain-derived neurotrophic factor genes in the mouse hippocampus and reduces anxiety-like behaviors. FASEB J. 2021, 35, e21767. [Google Scholar] [CrossRef]
- Cechinel, L.R.; Basso, C.G.; Bertoldi, K.; Schallenberger, B.; de Meireles, L.C.F.; Siqueira, I.R. Treadmill exercise induces age and protocol-dependent epigenetic changes in prefrontal cortex of Wistar rats. Behav. Brain Res. 2016, 313, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, F.; Zhuang, Y.; Feng, J.; Ying, Z.; Fan, G. Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur. J. Neurosci. 2011, 33, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. eLife 2016, 5, e15092. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Li, Y.; Dai, Y.; Li, L.; Lv, G.; Chen, I.; Wang, B. MiR-140/BDNF axis regulates normal human astrocyte proliferation and LPS-induced IL-6 and TNF-α secretion. Biomed. Pharmacother. 2017, 91, 899–905. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, S.; Li, Z.; Zhou, J. MicroRNA-211/BDNF axis regulates LPS-induced proliferation of normal human astrocyte through PI3K/AKT pathway. Biosci. Rep. 2017, 37, BSR20170755. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Bin, H.; Chen, W. Inhibition of microRNA-103a inhibits the activation of astrocytes in hippocampus tissues and improves the pathological injury of neurons of epilepsy rats by regulating BDNF. Cancer Cell Int. 2019, 19, 109. [Google Scholar] [CrossRef]
- Bao, T.-H.; Miao, W.; Han, J.-H.; Yin, M.; Yan, Y.; Wang, W.-W.; Zhu, Y.-H. Spontaneous Running Wheel Improves Cognitive Functions of Mouse Associated with miRNA Expressional Alteration in Hippocampus Following Traumatic Brain Injury. J. Mol. Neurosci. 2014, 54, 622–629. [Google Scholar] [CrossRef]
- Mishra, A.; Kohli, S.; Dua, S.; Thinlas, T.; Mohammad, G.; Pasha, M.A.Q. Genetic differences and aberrant methylation in the apelin system predict the risk of high-altitude pulmonary edema. Proc. Natl. Acad. Sci. USA 2015, 112, 6134–6139. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.N.; Dye, J.A.; Schladweiler, M.C.; Richards, J.H.; Ledbetter, A.D.; Stewart, E.; Kodavanti, U.P. Acute inhalation of ozone induces DNA methylation of apelin in lungs of Long-Evans rats. Inhal. Toxicol. 2018, 30, 178–186. [Google Scholar] [CrossRef]
- Keleher, M.R.; Zaidi, S.; Shah, S.; Oakley, M.E.; Pavlatos, C.; El Idrissi, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS ONE 2018, 13, e0192606. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Zeng, Z.-C.; Xi, M.; Wan, S.; Hua, W.; Liu, Y.-L.; Zhou, Y.-L.; Luo, H.-W.; Jiang, F.-N.; Zhong, W.-D. Dysregulated microRNA-224/apelin axis associated with aggressive progression and poor prognosis in patients with prostate cancer. Hum. Pathol. 2015, 46, 295–303. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, M.; Du, Y.; Liu, Y.; Zhao, G.; Ye, L.; Li, Q.; Li, H.; Wang, X.; Liu, X.; et al. MicroRNA-195 suppresses the progression of lung adenocarcinoma by directly targeting apelin. Thorac. Cancer 2019, 10, 1419–1430. [Google Scholar] [CrossRef]
- Liao, Y.-C.; Wang, Y.-S.; Hsi, E.; Chang, M.-H.; You, Y.-Z.; Juo, S.-H.H. MicroRNA-765 influences arterial stiffness through modulating apelin expression. Mol. Cell. Endocrinol. 2015, 411, 11–19. [Google Scholar] [CrossRef]
- Yang, M.; Song, J.-J.; Yang, X.-C.; Zhong, G.-Z.; Zhong, J.-C. MiRNA-122-5p inhibitor abolishes angiotensin II–mediated loss of autophagy and promotion of apoptosis in rat cardiofibroblasts by modulation of the apelin-AMPK-mTOR signaling. In Vitro Cell. Dev. Biol. Anim. 2022, 58, 136–148. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, Z.; Li, C.; Song, Y.; Wang, Y.; Bo, H.; Zhang, Y. Impact of Exercise and Aging on Mitochondrial Homeostasis in Skeletal Muscle: Roles of ROS and Epigenetics. Cells 2022, 11, 2086. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11132086
Li J, Wang Z, Li C, Song Y, Wang Y, Bo H, Zhang Y. Impact of Exercise and Aging on Mitochondrial Homeostasis in Skeletal Muscle: Roles of ROS and Epigenetics. Cells. 2022; 11(13):2086. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11132086
Chicago/Turabian StyleLi, Jialin, Zhe Wang, Can Li, Yu Song, Yan Wang, Hai Bo, and Yong Zhang. 2022. "Impact of Exercise and Aging on Mitochondrial Homeostasis in Skeletal Muscle: Roles of ROS and Epigenetics" Cells 11, no. 13: 2086. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11132086