Targeting Cellular Metabolism in Acute Myeloid Leukemia and the Role of Patient Heterogeneity

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Human Cells
2.1.1. Primary Human AML Cells
2.1.2. Primary Umbilical Cord Blood (UCB) Cells
2.2. Reagents
2.3. Analysis of Cell Proliferation, Viability and Constitutive Release of Mediators
2.3.1. AML Cell Proliferation
2.3.2. Cocultures of Human Mesenchymal Stem Cells (MSCs) and AML Cells
2.3.3. Viability
2.3.4. Soluble Mediator Release
2.4. Mutational Profiling, Global Gene Expression Profiling and Proteomic Analysis
2.4.1. Mutational Profiling
2.4.2. Global Gene Expression Profiling
2.4.3. Proteomic Analyses
2.5. Statistical Analyses
3. Results
3.1. Dose-Response Screening Studies of AML Cell Proliferation after Exposure to Metabolic Inhibitors
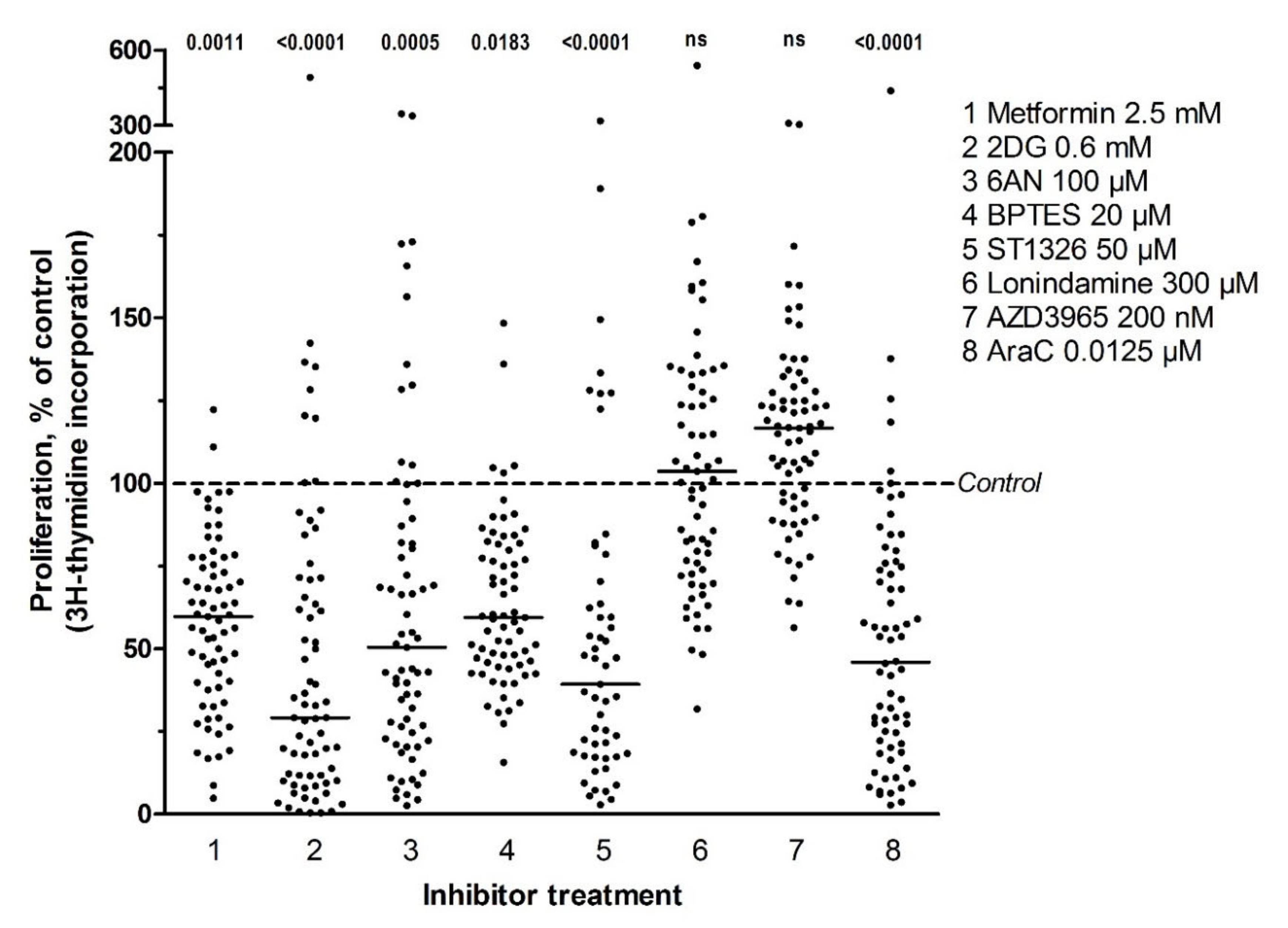
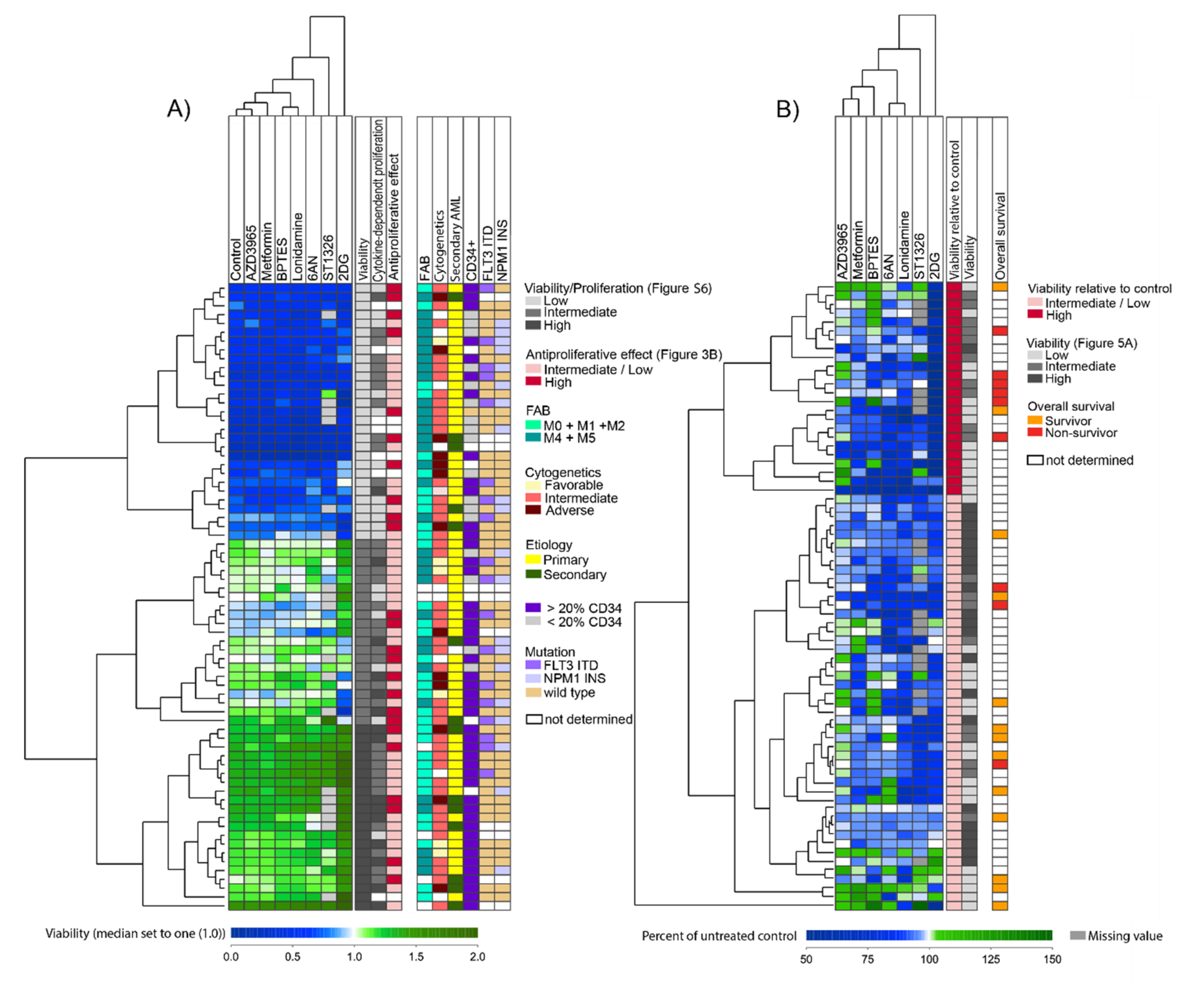
3.2. Several Metabolic Inhibitors Have Antiproliferative Effects on Primary AML Cells, with no Association with Secondary AML, AML cell Differentiation, Karyotype, FLT3 or NPM1 Mutations, or Patient Survival
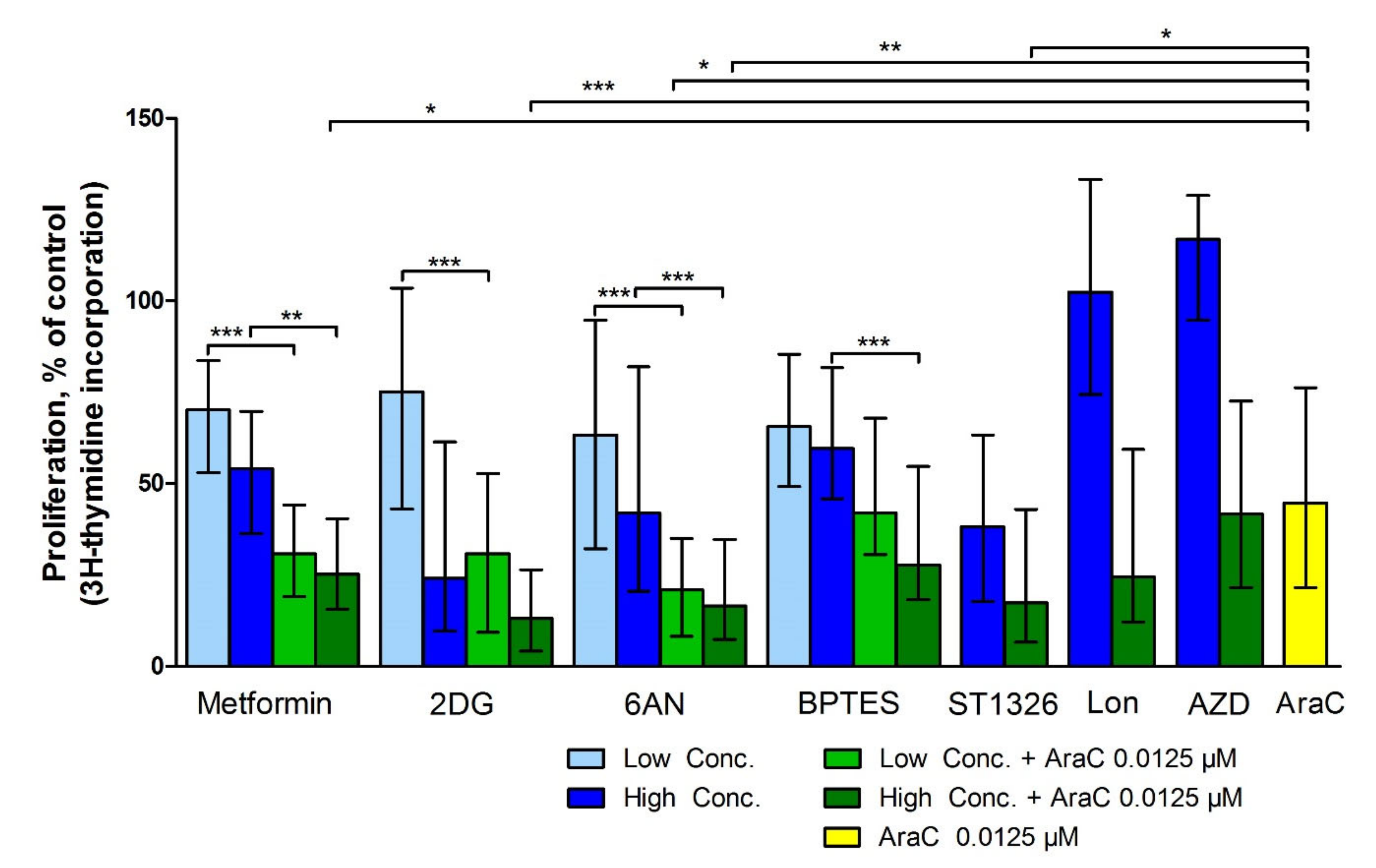
3.3. Metabolic Inhibitors Have Additive Antiproliferative Effects when Combined with AraC
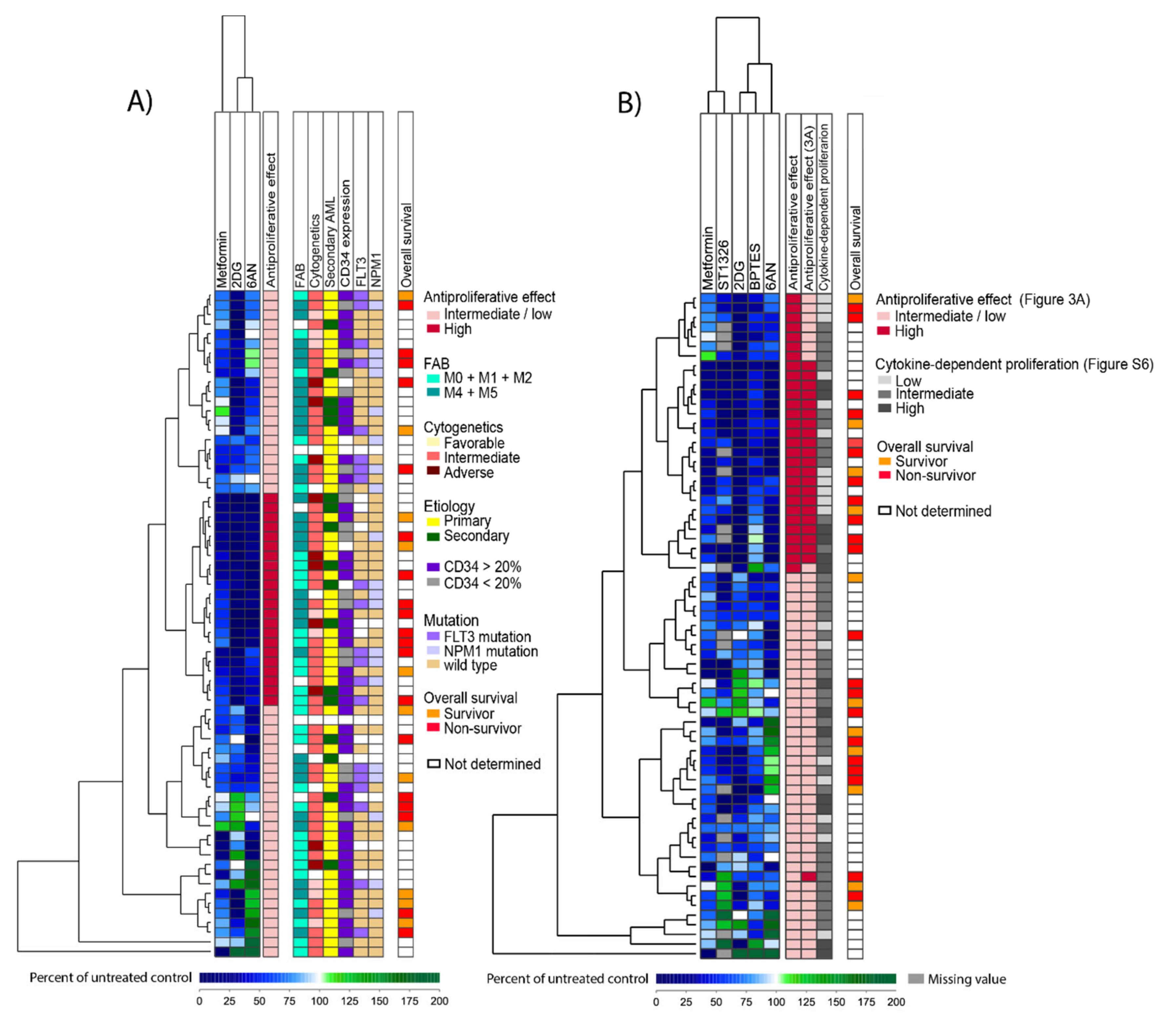
3.4. The Antiproliferative Effect of Metabolic Inhibitors Differs between Patients and a Subset of Patients Show Increased Susceptibility to Several Inhibitors
3.5. A Proteomic Comparison of Patient Samples Showing Either Generally Strong or Weak Antiproliferative Effects after Treatment with Metabolic Inhibitors
3.6. A Comparison of Global Gene Expression Profiles for AML Samples that Differ in Their General Susceptibility toward Metabolic Inhibitors
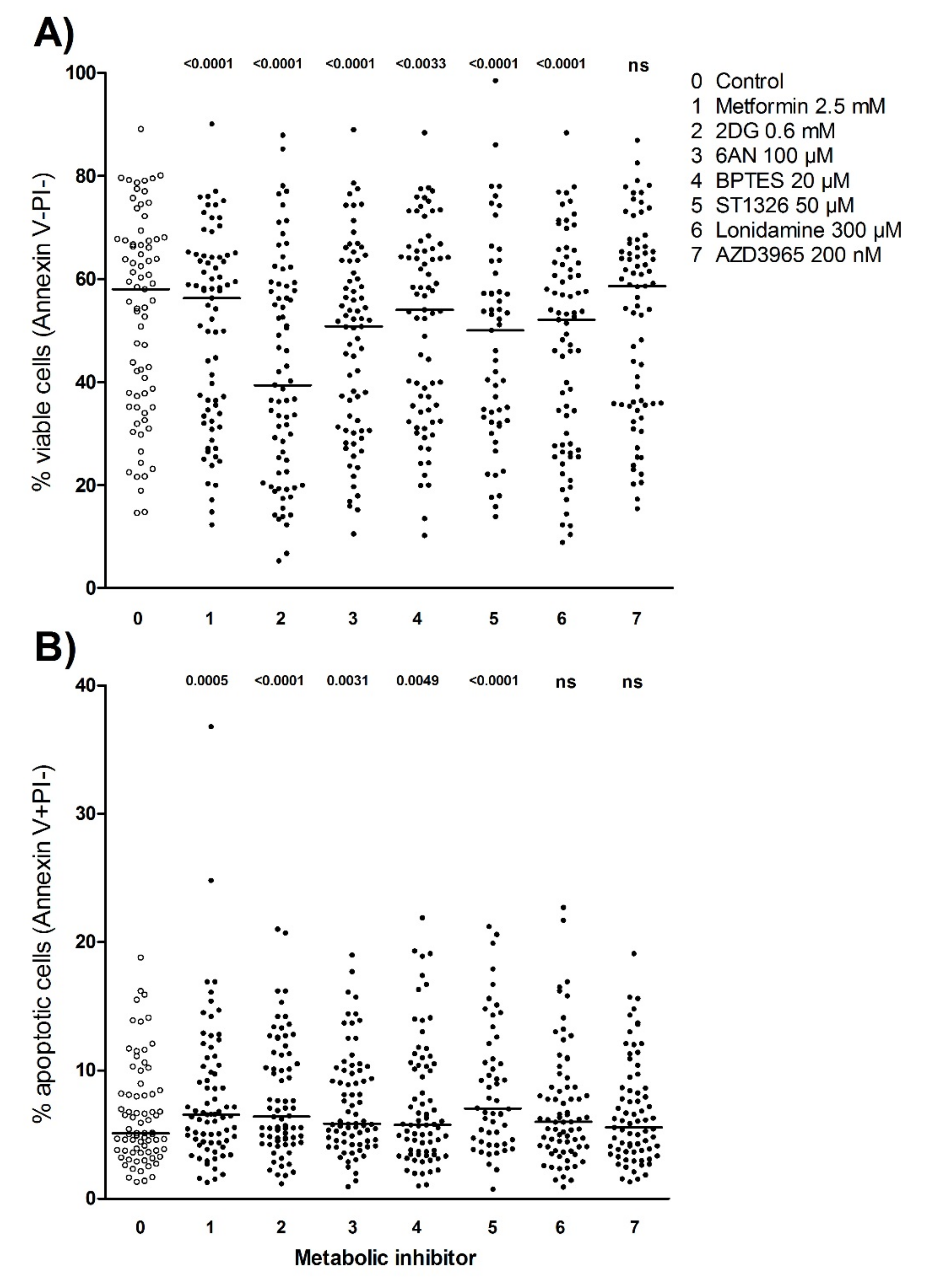
3.7. Metabolic Inhibitors Decrease AML Cell Viability through Proapoptotic Effects
3.8. Identification of Patient Subsets Based on Effects of Metabolic Inhibitors on AML Cell Viability
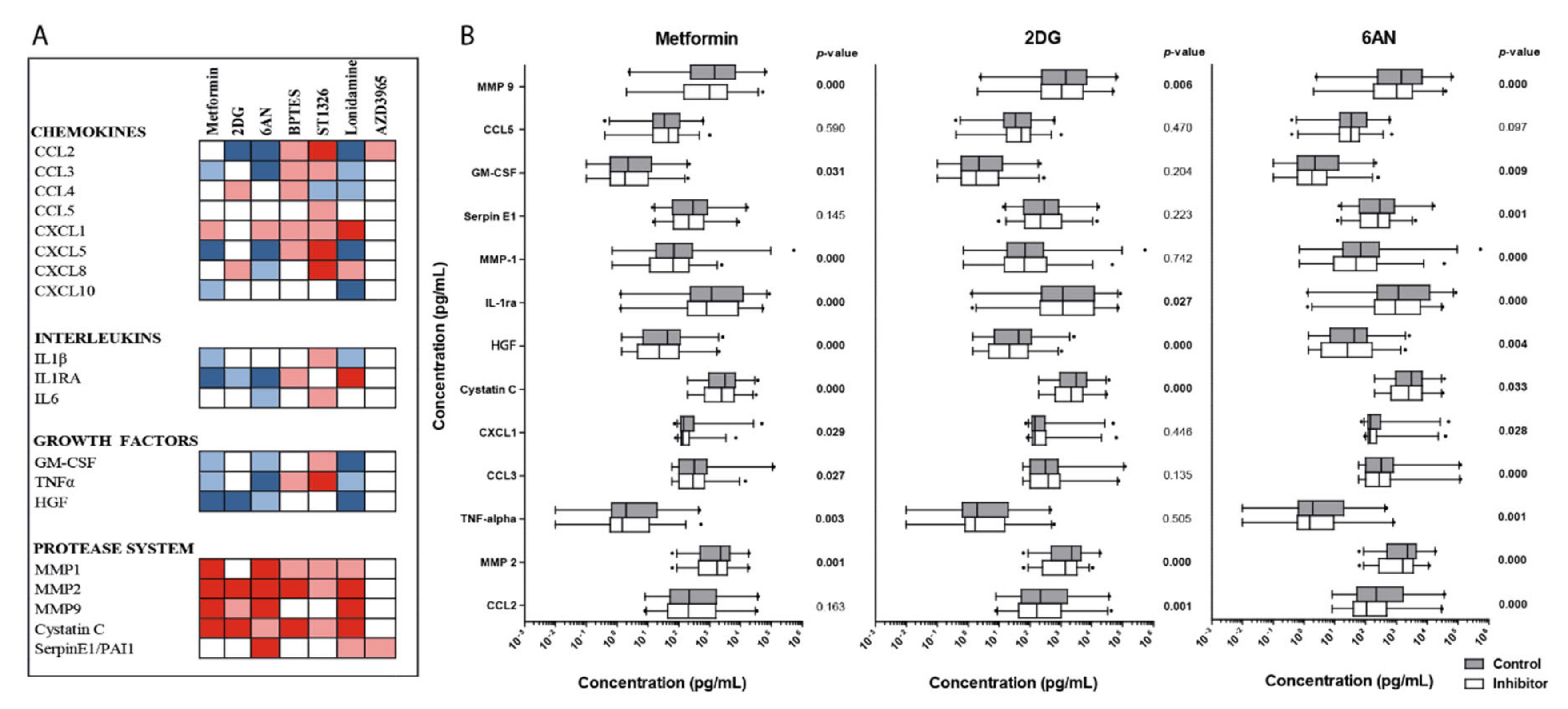
3.9. Effects of Metabolic Inhibitors on the Constitutive Release of Soluble Mediators By Primary AML Cells
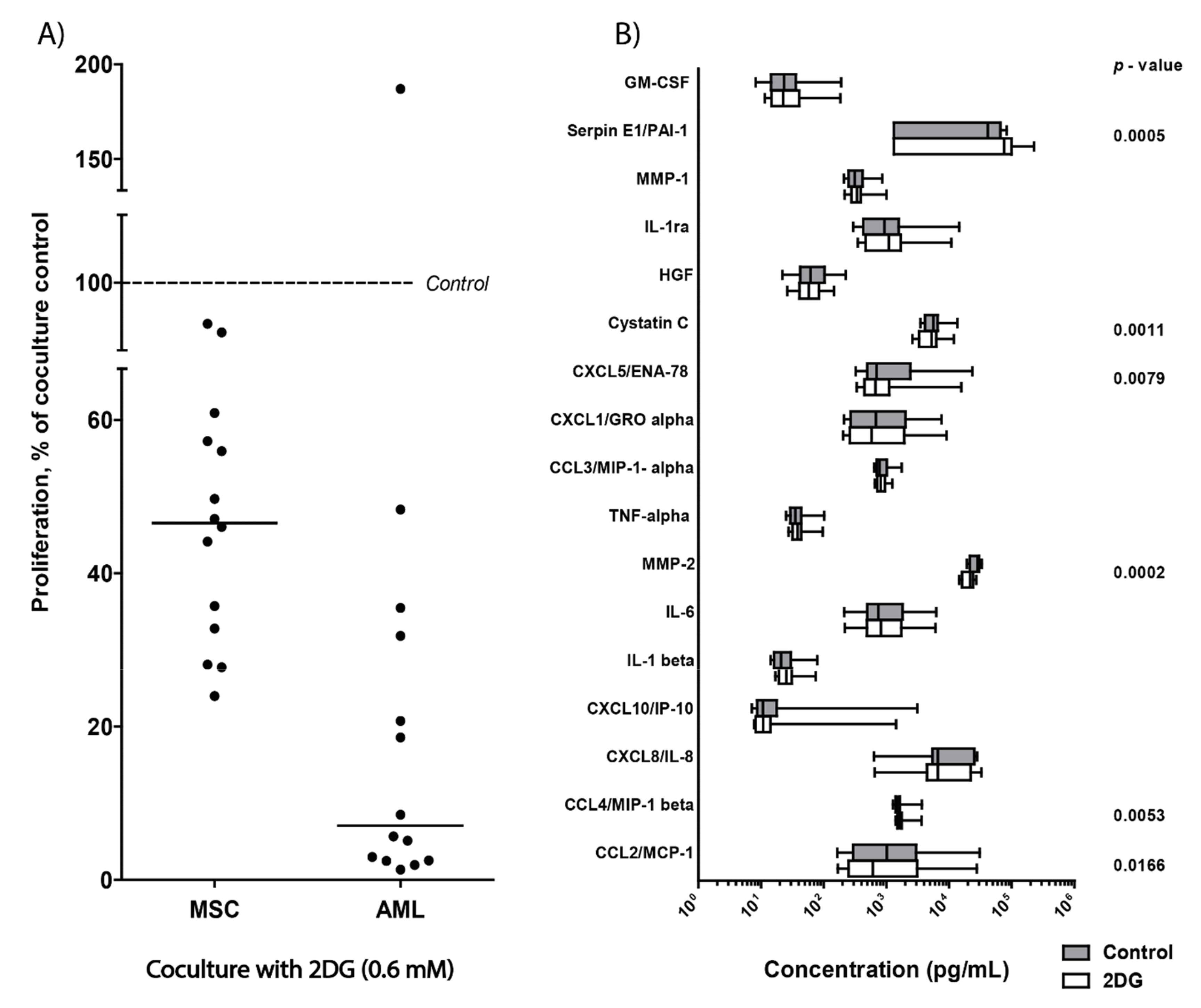
3.10. 2DG Has an Antiproliferative Effect on AML Cells Even in the Presence of AML-Supporting MSCs
3.11. Dose-Response Studies of Umbilical Cord Blood Cells Treated with Metabolic Inhibitors, Effects on Proliferation and Cell Viability
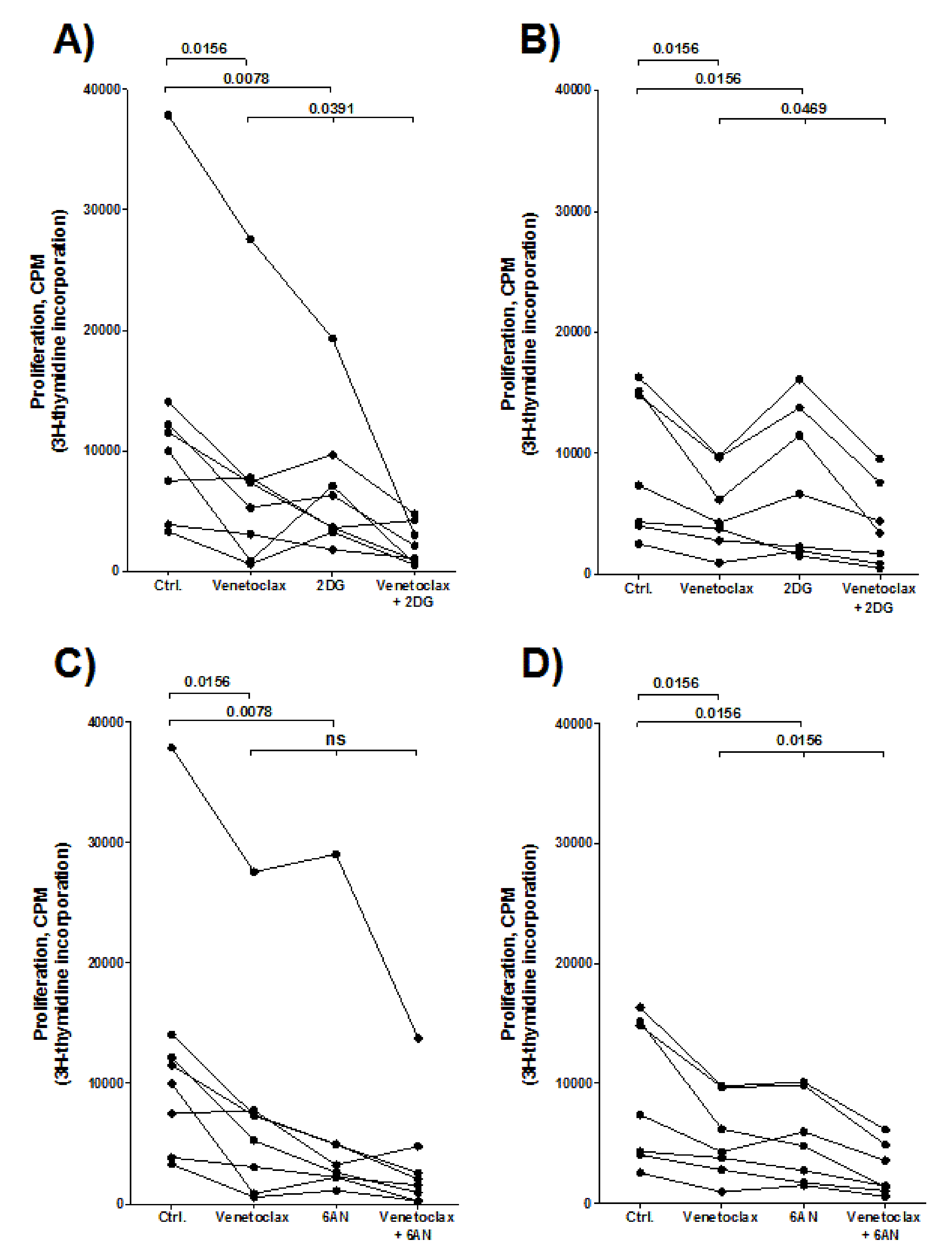
3.12. Combination Treatment of Venetoclax and a Metabolic Inhibitor (2DG or 6AN) has a Stronger Antiproliferative Effect on AML Cells than Venetoclax or the Metabolic Inhibitor Alone
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hospital, M.-A.; Green, A.S.; Maciel, T.; Moura, I.C.; Leung, A.Y.; Bouscary, D.; Tamburini, J. FLT3 inhibitors: Clinical potential in acute myeloid leukemia. OncoTargets Ther. 2017, 10, 607–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuani, L.; Sabatier, M.; Sarry, J.-E. Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Boil. 2019, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in The Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Hanahan, D.; A Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Nepstad, I.; Reikvam, H.; Brenner, A.; Bruserud, Ø.; Hatfield, K.J. Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. Int. J. Mol. Sci. 2018, 19, 382. [Google Scholar] [CrossRef] [Green Version]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [Green Version]
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.-E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, K.M.; Dewhirst, M.W. Tumor metabolism of lactate: The influence and therapeutic potential for MCT and CD147 regulation. Futur. Oncol. 2010, 6, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef]
- Chapuis, N.; Poulain, L.; Birsen, R.; Tamburini, J.; Bouscary, D. Rationale for Targeting Deregulated Metabolic Pathways as a Therapeutic Strategy in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Bruserud, Ø.; Gjertsen, B.T.; Von Volkman, H.L. In VitroCulture of Human Acute Myelogenous Leukemia (AML) Cells in Serum-Free Media: Studies of Native AML Blasts and AML Cell Lines. J. Hematotherapy 2000, 9, 923–932. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Hovland, R.; Wergeland, L.; Huang, T.-S.; Gjertsen, B.T. Flt3-mediated signaling in human acute myelogenous leukemia (AML) blasts: A functional characterization of Flt3-ligand effects in AML cell populations with and without genetic Flt3 abnormalities. Haematologica 2003, 88, 416–428. [Google Scholar]
- Bruserud, Ø.; Ryningen, A.; Wergeland, L.; Glenjen, N.I.; Gjertsen, B.T. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 2004, 89, 391–402. [Google Scholar]
- Stapnes, C.; Døskeland, A.P.; Hatfield, K.; Ersvær, E.; Ryningen, A.; Lorens, J.B.; Gjertsen, B.T.; Bruserud, Ø. The proteasome inhibitors bortezomib and PR-171 have antiproliferative and proapoptotic effects on primary human acute myeloid leukaemia cells. Br. J. Haematol. 2007, 136, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.; Nepstad, I.; Bruserud, Ø. Mesenchymal Stem Cells Support Survival and Proliferation of Primary Human Acute Myeloid Leukemia Cells through Heterogeneous Molecular Mechanisms. Front. Immunol. 2017, 8, 1063. [Google Scholar] [CrossRef] [Green Version]
- Ryningen, A.; Ersvær, E.; Øyan, A.M.; Kalland, K.-H.; Vintermyr, O.K.; Gjertsen, B.T.; Bruserud, Ø. Stress-induced in vitro apoptosis of native human acute myelogenous leukemia (AML) cells shows a wide variation between patients and is associated with low BCL-2:Bax ratio and low levels of heat shock protein 70 and 90. Leuk. Res. 2006, 30, 1531–1540. [Google Scholar] [CrossRef]
- Reikvam, H.; Hovland, R.; Forthun, R.B.; Erdal, S.; Gjertsen, B.T.; Fredly, H.; Bruserud, Ø. Disease-stabilizing treatment based on all-trans retinoic acid and valproic acid in acute myeloid leukemia - identification of responders by gene expression profiling of pretreatment leukemic cells. BMC Cancer 2017, 17, 630. [Google Scholar] [CrossRef] [Green Version]
- Stavrum, A.-K.; Petersen, K.; Jonassen, I.; Dysvik, B. Analysis of Gene-Expression Data Using J-Express. Curr. Protoc. Bioinform. 2008, 21, 7.3.1. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2018, 47, D419–D426. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, Ø.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Boil. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef] [Green Version]
- Reikvam, H.; Aasebø, E.; Brenner, A.; Bartaula-Brevik, S.; Grønningsæter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, Ø. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.V.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. New Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; DeBose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2013, 4, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Herst, P.; Howman, R.A.; Neeson, P.J.; Berridge, M.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Boil. 2010, 89, 51–55. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Gjertsen, B.T.; Foss, B.; Huang, T.-S.; Tjønnfjord, G.; Ernst, P. New Strategies in the Treatment of Acute Myelogenous Leukemia (AML): In Vitro Culture of AML Cells-The Present Use in Experimental Studies and the Possible Importance for Future Therapeutic Approaches. STEM CELLS 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Gjertsen, B.T.; Øyan, A.M.; Marzolf, B.; Hovland, R.; Gausdal, G.; Døskeland, S.O.; Dimitrov, K.; Golden, A.; Kalland, K.-H.; Hood, L.; et al. Analysis of Acute Myelogenous Leukemia: Preparation of Samples for Genomic and Proteomic Analyses. J. Hematotherapy 2002, 11, 469–481. [Google Scholar] [CrossRef]
- Sasnoor, L.M.; Kale, V.P.; Limaye, L. A combination of catalase and trehalose as additives to conventional freezing medium results in improved cryoprotection of human hematopoietic cells with reference to in vitro migration and adhesion properties. Transfusion 2005, 45, 622–633. [Google Scholar] [CrossRef]
- Griffiths, M.; Sundaram, H. Drug Design and Testing: Profiling of Antiproliferative Agents for Cancer Therapy Using a Cell-Based Methyl-[3H]-Thymidine Incorporation Assay. Adv. Struct. Saf. Stud. 2011, 731, 451–465. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.; Xuan, L.; Huang, G.-N.; Song, X.-L.; Liu, Q. HIF-1α and GLUT1 Gene Expression is Associated with Chemoresistance of Acute Myeloid Leukemia. Asian Pac. J. Cancer Prev. 2014, 15, 1823–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Li, M.; Xu, X.; Xuan, L.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salani, B.; Marini, C.; Del Rio, A.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin Impairs Glucose Consumption and Survival in Calu-1 Cells by Direct Inhibition of Hexokinase-II. Sci. Rep. 2013, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Salani, B.; Massollo, M.; Amaro, A.; Esposito, A.I.; Orengo, A.M.; Capitanio, S.; Emionite, L.; Riondato, M.; Bottoni, G.; et al. Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle 2013, 12, 3490–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.-S.; Árnadóttir, S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Martin, D.S.; Xu, R.-H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, F. Venetoclax plus hypomethylating agents or low-dose cytarabine in acute myeloid leukemia: All that glitters is gold? Blood Cancer J. 2020, 10, 1–4. [Google Scholar] [CrossRef]
- Lou, Y.; Shao, L.; Mao, L.; Lu, Y.; Ma, Y.; Fan, C.; Jiang, H.; Li, J.; Jin, J. Efficacy and predictive factors of venetoclax combined with azacitidine as salvage therapy in advanced acute myeloid leukemia patients: A multicenter retrospective study. Leuk. Res. 2020, 91, 106317. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; A Riemondy, K.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Xu, X. Knockdown of LncRNA-UCA1 suppresses chemoresistance of pediatric AML by inhibiting glycolysis through the microRNA-125a/hexokinase 2 pathway. J. Cell. Biochem. 2018, 119, 6296–6308. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.-Q.; Zhan, G.; Huang, A.; Sun, Y.; Wen, S.; Yang, J.; Lu, W.-H.; Xu, R.-H.; Li, J.; Li, Y.; et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 2017, 31, 2143–2150. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Pérez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.; et al. Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.; Nemkov, T.; Reisz, J.A.; Zaberezhnyy, V.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 2018, 58, 52–58. [Google Scholar] [CrossRef]
- Brattås, M.K.; Reikvam, H.; Tvedt, T.H.A.; Bruserud, Ø. Precision medicine for TP53-mutated acute myeloid leukemia. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 263–274. [Google Scholar] [CrossRef]
- Kheniser, K.; Kashyap, S.R.; Kasumov, T. A systematic review: The appraisal of the effects of metformin on lipoprotein modification and function. Obes. Sci. Pr. 2019, 5, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Grønningsæter, I.S.; Fredly, H.; Gjertsen, B.T.; Hatfield, K.J.; Bruserud, Ø. Systemic Metabolomic Profiling of Acute Myeloid Leukemia Patients before and During Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid, Valproic Acid, and Low-Dose Chemotherapy. Cells 2019, 8, 1229. [Google Scholar] [CrossRef] [Green Version]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Niraula, S.; Dowling, R.J.O.; Ennis, M.; Chang, M.C.; Done, S.J.; Hood, N.; Escallon, J.; Leong, W.L.; McCready, D.R.; Reedijk, M.; et al. Metformin in early breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat. 2012, 135, 821–830. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; DiPaola, R.S.; Stein, M.N.; Lima, C.M.R.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 71, 523–530. [Google Scholar] [CrossRef]
- Perlia, C.P.; Kofman, S.; Sky-Peck, H.; Taylor, S.G. 3rd Clinical use of 6-aminonicotinamide in patients with disseminated neoplastic disease. Cancer 1961, 14, 644–648. [Google Scholar] [CrossRef]
- Holubarsch, C.J.F.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Della Cuna, G.R.; Pedrazzoli, P. Toxicity and clinical tolerance of lonidamine. Semin. Oncol. 1991, 18, 18–22. [Google Scholar]
- Oudard, S.; Carpentier, A.; Banu, E.; Fauchon, F.; Celerier, D.; Poupon, M.F.; Dutrillaux, B.; Andrieu, J.M.; Delattre, J.Y. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J. Neuro-Oncol. 2003, 63, 81–86. [Google Scholar] [CrossRef]
- Halford, S.E.R.; Jones, P.; Wedge, S.; Hirschberg, S.; Katugampola, S.; Veal, G.J.; Payne, G.; Bacon, C.; Potter, S.; Griffin, M.; et al. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J. Clin. Oncol. 2017, 35, 2516. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLINICAL CHARACTERISTICS | |||||
|---|---|---|---|---|---|
| Gender (n) | Age (years) | ||||
| Female Male | 30 51 | Median Range | 67.5 17–87 | ||
| Relapse or previous hematological disease | Survival* | ||||
| AML relapse MDS CMML PV Li-Fraumeni syndrome | 5 11 2 1 1 | Yes No Not relevant | 15 24 42 | ||
| AML CELL DIFFERENTIATION | |||||
| FAB classification | CD34 expression | ||||
| M0/M1/M2 M4/M5 nd | 38 36 7 | Negative (<20%) Positive (≥20%) nd | 22 52 7 | ||
| GENETIC ABNORMALITIES | |||||
| Cytogenetics** | FLT3 | NPM-1 | |||
| Favorable Intermediate Adverse nd | 10 48 14 9 | wt ITD TKD ITD/TKD nd | 45 22 2 3 9 | wt INS nd | 45 28 8 |
| Metabolic Inhibitor | Molecular Target—Main Metabolic Pathway Affected |
|---|---|
| AZD3965 | Inhibits glycolysis; a selective inhibitor of monocarboxylate transporter 1 (MCT1) that regulates lactate transport across the plasma membrane. |
| Metformin | Inhibits oxidative phosphorylation (OXPHOS) and fatty acid metabolism; inhibits hexokinase activity, activates AMP-activated protein kinase (AMPK) and indirectly inhibits mammalian target of rapamycin (mTOR). |
| 2DG | Inhibits glycolysis; inhibits the rate-limiting enzyme hexokinase. |
| Lonidamine | Inhibits glycolysis and OXPHOS through multisite effects, including inhibition of hexokinase II, MCT1, the mitochondrial pyruvate carrier, the electron transport chain and alters mitochondrial permeability. |
| 6AN | Inhibits the pentose phosphate pathway (PPP) which is a main source of NADPH and ribose-5 phosphate. |
| BPTES | Inhibits glutaminase activity, that is the conversion of glutamine to glutamate. |
| ST1326 | Inhibits carnetyl palmitoyl transferase-1 (CPT-1), the rate-limiting step of fatty acid oxidation (FAO). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grønningsæter, I.S.; Reikvam, H.; Aasebø, E.; Bartaula-Brevik, S.; Tvedt, T.H.; Bruserud, Ø.; Hatfield, K.J. Targeting Cellular Metabolism in Acute Myeloid Leukemia and the Role of Patient Heterogeneity. Cells 2020, 9, 1155. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051155
Grønningsæter IS, Reikvam H, Aasebø E, Bartaula-Brevik S, Tvedt TH, Bruserud Ø, Hatfield KJ. Targeting Cellular Metabolism in Acute Myeloid Leukemia and the Role of Patient Heterogeneity. Cells. 2020; 9(5):1155. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051155
Chicago/Turabian StyleGrønningsæter, Ida Sofie, Håkon Reikvam, Elise Aasebø, Sushma Bartaula-Brevik, Tor Henrik Tvedt, Øystein Bruserud, and Kimberley Joanne Hatfield. 2020. "Targeting Cellular Metabolism in Acute Myeloid Leukemia and the Role of Patient Heterogeneity" Cells 9, no. 5: 1155. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051155