Novel PET Biomarkers to Disentangle Molecular Pathways across Age-Related Neurodegenerative Diseases



1
Neurodegeneration Imaging Group, University of Exeter Medical School, London W12 0BZ, UK
2
Invicro, Centre for Imaging Sciences, Hammersmith Hospital, London W12 0NN, UK
3
Department of Neuroimaging, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London SE5 8AF, UK
4
School of Public Health, Imperial College London, London W2 1PG, UK
5
Public Health Directorate, Imperial College NHS Healthcare Trust, London W6 8RF, UK
*
Author to whom correspondence should be addressed.
Cells 2020, 9(12), 2581; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122581
Submission received: 28 October 2020
/
Revised: 25 November 2020
/
Accepted: 28 November 2020
/
Published: 2 December 2020
(This article belongs to the Collection Novel Biomarkers for Alzheimer’s Disease and Other Neurodegenerative Diseases)
Abstract
:There is a need to disentangle the etiological puzzle of age-related neurodegenerative diseases, whose clinical phenotypes arise from known, and as yet unknown, pathways that can act distinctly or in concert. Enhanced sub-phenotyping and the identification of in vivo biomarker-driven signature profiles could improve the stratification of patients into clinical trials and, potentially, help to drive the treatment landscape towards the precision medicine paradigm. The rapidly growing field of neuroimaging offers valuable tools to investigate disease pathophysiology and molecular pathways in humans, with the potential to capture the whole disease course starting from preclinical stages. Positron emission tomography (PET) combines the advantages of a versatile imaging technique with the ability to quantify, to nanomolar sensitivity, molecular targets in vivo. This review will discuss current research and available imaging biomarkers evaluating dysregulation of the main molecular pathways across age-related neurodegenerative diseases. The molecular pathways focused on in this review involve mitochondrial dysfunction and energy dysregulation; neuroinflammation; protein misfolding; aggregation and the concepts of pathobiology, synaptic dysfunction, neurotransmitter dysregulation and dysfunction of the glymphatic system. The use of PET imaging to dissect these molecular pathways and the potential to aid sub-phenotyping will be discussed, with a focus on novel PET biomarkers.
1. Introduction
It is widely accepted that age-related neurodegenerative diseases are increasingly becoming a global public health concern—in particular, Alzheimer’s disease (AD) and other late-onset dementias (LOD), with widespread socioeconomic and healthcare impacts worldwide. The increasing burden of age-related diseases is mainly due to the ageing world population and the unprecedented shift in aging demographics of individuals over 60 years of age, which is predicted to rise to two billion in 2050 [1]. Age-related neurodegenerative diseases encompass a spectrum of complex and heterogenous diseases, including AD, Parkinson’s disease (PD), Parkinson’s disease Dementia (PDD), Dementia with Lewy Bodies (DLB), the recently identified dementia form of “Limbic-predominant Age-related TDP-43 Encephalopathy (LATE)”, late-onset forms of Fronto-Temporal Dementia (FTD) and of Amyotrophic Lateral Sclerosis (ALS), as well as parkinsonian plus syndromes, such as Corticobasal Syndrome (CBS), Progressive Supranuclear Palsy (PSP) and Multiple System Atrophy (MSA). Unlike other public health challenges, such as cancer, which have seen the recent development of effective disease modifying treatments, therapies for age-related neurodegenerative diseases remain ineffective to modify the disease course, with most therapies only providing some symptomatic relief.
The majority of research across age-related neurodegenerative diseases is built upon the clinicopathological nosology model [2], whereby a specific clinical phenotype is studied aiming to unlock the underlying pathology, traditionally through post-mortem investigations and, more recently, through in vivo studies, using imaging and other biomarkers that reflect key pathological changes. Variations across diseases have been attributed to the selective vulnerability of specific neuronal subtypes to disease pathology that subsequently determine the clinical phenotypic expression. However, the majority of age-related neurodegenerative diseases are complex in nature, resulting from poorly understood interactions between genomic, environmental and lifestyle factors, across the life course, and harbor multiple pathologies; as a result, their clinical presentations can have distinct, as well as overlapping, features occurring at different levels and timepoints [3,4,5]. The concept that neuronal networks, rather than neuronal subtypes, could underlie differences in the clinical phenotype and the susceptibility of individuals to different neurodegenerative diseases has gained increasing interest over the last decade, aiming to unlock the paradigm of age-related neurodegenerative diseases [4,6,7,8,9]. In 2018, the National Institute on Aging and the Alzheimer’s Association proposed the Research Framework classification of AD to better define the diagnosis of AD, across a disease continuum from preclinical to severe clinical stages from other LOD forms, based on the in vivo AT(N) biomarker signature, corresponding to the three landmark pathological features of increased Amyloid and Tau burden, associated with a significant loss of volume and neurodegeneration [10,11]. While this classification is a step towards the application of biological signature profiles, consideration should be given to the use of a binary classification model for continuous variables, based on a predetermined threshold, as a predictive or diagnostic tool in clinical trials, especially when using a clinical phenotype, such as cognitive decline or dementia, as the primary outcome measure [12].
Almost all age-related neurodegenerative diseases can be classified into sporadic or familial forms. The discovery of fully penetrant genetic mutations in several familial neurodegenerative diseases has allowed for the investigation of the early disease pathology prior to the clinical manifestations, in these familial forms, prior to the manifestation of clinical symptoms that could help to unlock causal pathways. In the nonfamilial sporadic forms, in addition to the genetic variants that are noncausative but can confer susceptibility to disease, there is a wide range of risk factors that may affect disease onset and development, including environmental factors and exposures across the life course, cardiovascular status and hypertension, obesity, diabetes, sleep disorders and a variety of factors related to brain biological aging, such as protein misfolding and aggregation, epigenetics and perturbations in DNA damage and repair. However, not all patients with all, or some, of these risk factors will develop symptoms and signs amounting to a clinical diagnosis within their lifetime. While the interaction between genetics and disease mechanisms is indeed complex and has not been fully elucidated, it has been postulated that unraveling the genetics of age-related neurodegenerative diseases might form the basis for sub-phenotyping and/or reclassification based on genotypic divergence aiming to drive forward the application of precision medicine [13].
There is a need to disentangle the etiological puzzle of age-related neurodegenerative diseases, whose clinical phenotypes arise from known, and as yet unknown, pathways that can act distinctly or in concert (Figure 1). Over the last 40 years, preclinical animal studies and post-mortem evaluations have unlocked a number of disease mechanisms and therapeutic targets, which showed promise to translate into novel therapies for age-related neurodegenerative diseases. In AD, the main causal hypotheses involved the amyloid cascade and the tau phosphorylation-propagation hypothesis. However, the majority of clinical trials targeting these mechanisms have failed to meet their primary endpoints [14,15,16]. The failure of clinical trials, across age-related neurodegenerative diseases, could be due to a number of reasons such as the late initiation of treatments in the disease course, poor target engagement or selection of the tested compound, suboptimal cohort stratification and the inability to reach the required effect sizes due to inadequate sample size and/or short follow-up periods [15]. Moreover, an inadequate appreciation of the complexity of disease etiology and pathophysiology can lead to an oversimplified mono-therapeutic approach [16].
While preclinical and post-mortem studies have, and will likely continue to, play a key role in the drug discovery process, as well as in understanding the underlying molecular mechanisms, considerations have to be given to their direct translation into humans [14,17]. For example, animal models of late-onset neurodegenerative diseases typically develop symptoms and die young, whereas in humans these diseases typically occur in late life [18]. Furthermore, post-mortem studies provide insights into pathological changes at a single timepoint (the very end stage of the disease), which can be contaminated from chronic drug treatments and other pathologies, making it difficult to disentangle whether the changes observed are a cause or consequence of neuronal death. The rapidly growing field of neuroimaging offers valuable tools to investigate disease pathophysiology and molecular pathways in vivo in humans, with the potential to capture the whole disease course. Positron emission tomography (PET) imaging combines the advantages of a versatile imaging technique with the ability to quantify, to nanomolar sensitivity, molecular targets, both in animals and in living humans. Magnetic resonance imaging (MRI) techniques can offer high spatial resolution and anatomical granularity with advanced acquisition protocols and analysis methodologies offering a platform to explore microstructural and functional connectivity, iron deposition, neuromelanin levels and neuro-hydrodynamics. Therefore, PET and MRI techniques are commonly employed in unison to extrapolate meaningful outcome measures reflecting molecular biology in vivo.
This review will discuss the dysregulation of the main molecular pathways, pathology and biological networks, highlighting where these are distinct and overlapping, across the spectrum of age-related neurodegenerative diseases. The use of molecular PET imaging to disentangle molecular pathways will be highlighted, focusing on novel biomarkers. Finally, the potential for biomarker-driven epigenomic, biological and clinical signatures to improve disease sub-phenotyping for the stratification of patients into future clinical trials towards the precision medicine icon will be discussed.
2. Dysregulation of Interlinked Molecular Pathways across Age-Related Neurodegenerative Diseases
While the temporal onset and the rate of progression can vary, clinical phenotypes, such as behavioral, cognitive, metabolic, nonmotor, primary motor and extrapyramidal, often overlap across different age-related neurodegenerative diseases (Figure 1). For example, patients with FTD can present with extrapyramidal symptoms similar to PD; AD patients can experience nonmotor symptoms such as sleep problems, which overlap with nonmotor symptoms observed in PD and parkinsonian plus syndromes, and patients with ALS can present with behavioral symptoms, such as apathy, which can overlap with FTD, parkinsonism plus syndromes and AD [19,20,21]. The pathogenesis and progression of age-related neurodegenerative diseases likely involves a dynamic interaction between various components and pathways at the genetic and pathological levels (Figure 1). Specific PET radioligands have been developed to target some of these molecular components, enabling the exploration of these pathways in vivo. There are a number of genotypic and molecular pathways that show varying degrees of overlap and crossover at various stages of disease etiology and progression. For example, while the clinical phenotype of three causative genes for FTD, C9orf72, MAPT and GRN, are associated with a similar behavioral variant FTD (bvFTD) presentation, the underlying protein pathology varies such that MAPT mutations are associated with tau pathology and C9orf72 and GRN mutations are associated with Tar-DNA-binding protein (TDP)-43 pathology [3]. Furthermore, a number of studies have unlocked genetic signatures that are common across different age-related neurodegenerative diseases. A meta-analysis of 1270 post-mortem brain tissue samples from AD, PD, ALS and Huntington’s disease (HD) patients identified shared gene expression signatures for 243 genes [22]. The common genes identified across these different diseases were related to functional pathways, including inflammation, synaptic signaling, metabolic dysfunction and oxidative stress. Moreover, while the causal role of epigenetics on age-related neurodegenerative diseases remains a topic of debate [23], similarities in the dysregulation of transcriptional networks and protein interaction networks have been reported [5].
It remains to be elucidated why, and how, pathologies diverge towards different clinical phenotypes and if there is a common causal mechanism that links the spectrum of age-related neurodegenerative diseases. The molecular nexopathies paradigm, introduced by Warren and colleagues, proposes that specific pathogenic proteins result in the disintegration of specific neural networks and multiple functional networks, which could give rise to phenotypic variations, as well as overlap between neurodegenerative diseases [4]. A deeper understanding of interlinked and distinctive molecular pathways, which drive pathological and clinical consequences, could provide novel therapeutic strategies. This section will highlight known overlapping, and distinct, molecular pathologies and pathways, focusing on the use of molecular PET imaging, in age-related neurodegenerative diseases (Figure 2).
3. Mitochondrial Dysfunction and Energy Dysregulation
There is an increasing body of literature implicating dysfunction of mitochondria and endoplasmic reticulum (ER) dynamics, energy metabolism and oxidative stress within the molecular paradigm of age-related neurodegenerative diseases [24,25,26,27,28,29]. Protein aggregation and deposition have been linked with mitochondrial dysfunction, disrupted mitochondrial transport, dysregulation of adenosine triphosphate (ATP) production, calcium imbalance and oxidative stress [28]. Furthermore, mitochondrial dysfunction can alter the energy supply to synapses, which could drive synaptic disconnection, contributing towards synaptic dysfunction and loss [30,31]. The identification of several genes, such as PINK-1, Parkin, TREM2, APOE and TOMM40 [32,33,34,35], which play key roles in the normal functioning of mitochondria has also highlighted the role of mitochondrial dysfunction in disease pathogenesis [36,37,38]. The temporal sequence of events and the exact interplay between mitochondria and ER dysfunction, oxidative stress, neuroinflammation and protein deposition remains to be fully elucidated. There are lines of evidence to support the accumulation of toxic proteins preceding and triggering mitochondrial and ER dysfunction [39,40,41]. Conversely, other evidence suggests that mitochondrial dysfunction and, consequently, oxidative stress and calcium imbalance, together with dysfunction of the ER, may lead to protein misfolding and the accumulation of toxic protein aggregates [42,43].
The development of novel PET radioligands, (18F)BCPP-EF, for mitochondrial complex 1 (MC1) and (11C)SA-4503 for sigma 1 receptor (σ1R) enables the in vivo investigation of mitochondrial and ER dysfunction (Figure 2A) in late-onset neurodegenerative and other diseases related to aging [44,45]. Sigma-1 receptors are expressed at the mitochondrion-associated ER membrane, where they regulate calcium signaling from the ER to the mitochondrion [46,47,48]. Sigma-1 receptors also display neuromodulator and neuroprotective properties, aiding protein folding and modulating synaptic neurotransmitter functions [46,49,50]. MC1 plays a fundamental role in cellular energy production, acting as the first rate limiting step of oxidative phosphorylation in the electron transport chain in mitochondria, as well as maintaining calcium homeostasis and regulating reactive oxygen species (ROS) levels [51,52]. The altered expression and dysfunction of σ1R and MC1 have been illustrated from post-mortem and preclinical studies in ALS, AD and PD [49,50,53,54,55,56,57].
Recently, σ1R and MC1 levels were investigated in a cohort of early de novo PD patients using (11C)SA-4503 and (18F)BCPP-EF PET, respectively [58]. Lower levels of σ1R and MC1 were observed at the baseline, but there were no significant cross sectional or longitudinal changes at 12-months follow-up. In another small cohort of moderate levodopa-treated PD patients, decreased striatal σ1R levels was reported [59,60], suggesting that the loss of σ1R might be more prominent in moderate-to-advanced disease stages. A combined (18F)BCPP-EF and (11C)PE2I PET preclinical study demonstrated that the striatal loss of MC1 correlated with the loss of presynaptic nigrostriatal dopaminergic neurons, supporting the interplay and colocalization of mitochondrial and synaptic dysfunction in a PD model [61]. Work is ongoing to investigate the role of σ1R and MC1 in AD, ALS, FTD and HD using (11C)SA-4503 and (18F)BCPP-EF PET, respectively, as part of the MIND-MAPS program (https://lp.invicro.com/mind-maps), which could help to provide a more comprehensive understanding of the mitochondrial-ER-synaptic complex, across the spectrum of age-related neurodegenerative diseases. Preliminary work suggests decreased MC1 density in AD [62] and FTD patients [63], with the loss of MC1 associated with global cognitive impairment across cohorts of age-related neurodegenerative cohorts [64]. Furthermore, preliminary findings indicate that σ1R density is increased in early AD, suggesting that this may represent a potential cellular response to stress that could subsequently decrease as the disease progresses [62]. Reduced (18F)BCPP-EF uptake has also been shown to correlate with increase tau deposition, using (11C)PPB3 PET, but not with amyloid-β, using (11C)PiB PET, or glucose metabolism, using (18F)FDG PET [65]. These preliminary findings could indicate that tau pathology precedes early mitochondria-related energy failure. However, these findings need to be further validated in larger, longitudinal studies. The temporal relationship between mitochondrial dysfunction, energy dysregulation and synaptic neuropathology warrants further investigation, as it may play a key role in the development of several age-related neurodegenerative diseases.
4. Immune Activation and Neuroinflammation
Neuroinflammation and alterations in the immune response have been linked with multiple pathological processes associated with age-related neurodegenerative diseases [66]. While there is evidence to implicate the role of the adaptive immune system [67,68], the majority of molecular PET imaging research to-date has focused on microglia and astrocytes as part of the innate immune system [69,70,71]. Stress factors, such as misfolded protein aggregates [72], could disrupt the tightly regulated balance between protective and detrimental effects of glial response. This imbalance could result in microglia and astroglia induced neurotoxicity via ROS, proinflammatory cytokines resulting in chronic neuroinflammation and glutamatergic excitotoxicity [73,74,75,76]. It has recently been reported that the crosstalk between microglia and astroglia activation may also contribute to dysregulation of the immune response following the accumulation of protein aggregates, mitochondrial dysfunction and progressive neuronal damage [77]. In ALS, it has been postulated that the shift in glial cells from neuroprotective to neurotoxic effect could contribute towards disease pathology [78]. The identification of genetic mutations in genes regulating microglial activation, such as TREM2 and CD33, in AD, FTD and PD [79,80,81] further supports the potential of a common causal role of a dysregulated immune response.
The majority of in vivo PET imaging studies investigating the role of neuroinflammation have focused on the use radioligands targeting the 18-kDa translocator protein (TSPO), expressed in the outer mitochondrial membrane and elevated in activated microglia (Figure 2B). A wide variety of TSPO ligands, such as (11C)PK1195, (11C)PBR28, (18F)DPA-714, (11C)DAA1106 and (11C)ER176, have been employed [82,83,84]. Increased binding of TSPO PET radioligands, interpreted as microglia activation, has been reported in PD [85,86,87,88], PDD [89], MSA [90], PSP [91], CBS [92], HD [93], AD [94,95], FTD [96,97] and ALS [98,99]. Increased (11C)PK1195 uptake has also been associated with higher amyloid-β burden in AD [100] and with reduced glucose metabolism in AD and PDD [101], suggesting an in vivo link between microglia activation, protein aggregation and energy dysregulation. Despite their wide application, there are a number of limitations to take into consideration when interpreting results for TSPO PET radioligands, such as the poor signal-to-noise ratio and high levels of nonspecific binding of the first-generation TSPO radioligands and the sensitivity to single-nucleotide polymorphisms in the TSPO gene for second-generation radioligands [100,102]. Third-generation TSPO radioligands, such as (11C)ER176 [82,83], which address some of these limitations warrant applications across age-related neurodegenerative diseases. However, the greatest challenge in the use of TSPO radioligands is not related to their imaging characteristics but the complicated biology underlying TSPO density changes. For example, TSPO radioligands cannot distinguish between different neuroprotective or neurotoxic isoforms of microglia, and more generally, there is an ongoing debate over the exact function and role of TSPO upregulation for the immune response.
The identification of reliable molecular targets for microglia activation beyond TSPO has been the focus for the development of the next generation of PET radioligands assessing neuroinflammatory biomarkers. Emerging PET radioligands that could provide key insights into inflammatory pathways include (11C)PS13 and (11C)MC1 for cyclooxygenase (COX)-1 and COX-2, respectively, and (11C)JN717 and (11C)SMW139 for the purinergic receptor P2X7 [103,104]. Recently, increased (18F)DPA714 PET in the absence of increased (11C)JN717 PET has been reported in ALS [99]. Studies are underway investigating P2X7 receptor in PD patients, using (11C)SMW139 PET (EudraCT Number: 2018-000405-23). The development of the PET radioligand (11C)BU99008 targeting imidazoline 2-binding sites (I2BS) expressed on activated astrocytes [105] has sparked the in vivo investigation of astroglial activation (Figure 2B) across several age-related neurodegenerative diseases [106,107,108,109]. Recently, astroglial activation, reflecting increased (11C)BU99008 binding, has been reported in early PD patients with decreased (11C)BU99008 binding, possibly reflecting that loss of astroglia function occurs in moderate-to-advanced PD disease stages and is associated with longer disease duration and higher global disease burden [108]. Preliminary findings show increased (11C)BU99008 PET in AD, with the highest levels in amyloid-β-positive AD patients [106,109], suggesting a role of astrogliosis in the pathophysiology of AD. Furthermore, preliminary work suggests astroglia activation is present also in PDD, suggesting a potential role in cognitive impairment in later stages of PD [107].
5. Protein Aggregates and the Concept of Pathobiology
A common feature across the spectrum of age-related neurodegenerative diseases is the presence of misfolded proteins. The type of protein pathology, as well as the temporal and spatial distribution, can vary between diseases that led to the early pathology-based classification of neurodegenerative diseases, according to the presence and spread of specific misfolded proteins. However, the presence of overlapping pathologies suggests a complex interplay between protein aggregates with disease etiopathogenesis, progression and clinical phenotypes. For example, in AD brains, α-synuclein has been reported to coexist with amyloid-β in senile plaques and in degenerating neuritis [110], and glial tau, neuronal tau and TDP-43 pathology, predominant pathology associated with FTD, limbic-predominant age-related TDP-43 encephalopathy (LATE) dementia and ALS, can also be present in AD and parkinsonian plus syndromes [3,111]. Furthermore, PET imaging has demonstrated that AD-related pathology, such as tau neurofibrillary tangles and amyloid-β plaques, can also coexist within Lewy bodies in PDD and DLB. In DLB, it has been shown that the pathological interplay between tau, amyloid-β and α-synuclein plays a role in the development of dementia [112,113,114,115,116,117], whilst amyloid-β brain accumulation is not typical in PDD [117,118]. Recently, the presence of APOE-ε4 and TOMM40-L alleles has been shown to be associated with the presence of AD-like pathology in DLB, while similar associations were not identified in PDD [33]. Across age-related neurodegenerative diseases, the chronology of mixed protein pathologies, together with multidimensional interactions between genetic and biological pathways, still remains obscure.
The last decades has witnessed the development and application of a range of PET radioligands to quantify amyloid-β and tau pathology in vivo (Figure 2C). The use of selective PET radioligands led to an eruption of literature describing the presence of protein pathology, the relevance to clinical phenotypes and the relationship with other molecular, structural and functional imaging markers across age-related neurodegenerative diseases [119,120,121,122]. Given the interplay between protein pathologies, it is likely that the development of novel PET radioligands to quantify Huntingtin, TDP-43 and α-synuclein in vivo is required to fully disentangle the relationship between the co-occurrence of complex protein pathologies and molecular pathways of neurodegeneration. Moreover, the ability to reliably measure the spectrum of protein pathology in the human brain would provide the ability to monitor novel multifaceted therapeutic approaches.
Amyloid PET imaging, using (11C)PiB, (18F)Florbetaben, (18F)Flutemetamol and (18F)Florbetapir, has been extensively employed to investigate disease specific patterns across age-related neurodegenerative diseases. Amyloid PET imaging can detect the presence of amyloid-β plaques prior to the onset of AD [123], predict cognitive decline in patients with amnestic mild cognitive impairment (MCI) [124,125] and aid the clinical diagnosis of AD [126]. However, amyloid PET does not appear to correlate strongly with cognitive decline once early AD is established [127]. Moreover, significant amyloid-β burden can also be detected in healthy aging individuals without dementia [128,129], suggesting that the presence of amyloid-β plaques alone may not be sufficient to drive cognitive decline. Increased amyloid PET uptake is associated with cognitive impairment in DLB patients [130], while PDD patients show mixed findings with some studies reporting increased amyloid-β levels associated with cognitive decline [114,116] and other studies reporting no relationship between amyloid-β deposition and cognitive impairment [118,130,131]. Therefore, further studies are warranted to untangle the relationship between amyloid-β and clinical phenotypes, as well as the interplay with co-occurrent protein pathologies in vivo.
PET studies employing tau ligands, such as (18F)Flortaucipir [132], have highlighted the association between tau pathology and cognitive performance across the AD continuum [133,134,135], PSP [136], CBS [137,138] and FTD [139] as well as tau co-pathology in Lewy body diseases [140]. A recent study supported the accuracy of (18F)Flortaucipir PET visual reads for predicting the presence of AD-like tau pathology at autopsy, suggesting the potential clinical use of (18F)Flortaucipir in the diagnosis of AD [141]. A main limitation of first-generation PET radioligands is the presence of off-target binding to neuromelanin and monoamine oxidase [142,143]. The second generation of tau PET radiotracers, including (18F)JNJ64349311 [144], (18F)APN-1607 ((18F)PM-PBB3) [145], (18F)MK-6240 [146,147,148], (18F)GTPI [149], (18F)PI-2620 [150] and (18F)RO-948 [151,152,153], offer promising in vivo tools to aid the differential diagnosis between AD and non-AD tau pathology and aid the development of novel pharmacotherapies. Recent findings from (18F)PI-2620 PET in AD and PSP illustrate tracer uptake in regions of known tau pathology, in line with post-mortem autoradiography findings [150,154]. Increased (18F)PI-2620 uptake within neocortical regions was associated with cognitive impairment in AD [150] and could aid the differential diagnosis of PSP [154]. Another recent study also illustrated that (18F)MK-6240 PET could differentiate between AD and FTD, with patterns of (18F)MK-6240 uptake in line with Braak’s histopathological staging of tau pathology in AD and negligible (18F)MK-6240 uptake in FTD supporting the specificity of this PET tracer for tau tangle conformations [148].
Multifaceted neuroimaging approaches, combining tau PET, amyloid-β PET or FDG PET (for glucose metabolism) with functional and structural MRI techniques, could potentially help to unlock network-based connectivity disease signatures. For example, spatial covariance mapping has been employed to derive network topography underlying clinical phenotypes such as cognitive impairments [155]. Disease-specific networks could also shed light on the relationship between genotypes, disease progression and clinical phenotypes [156]. Recently, the combination of tau PET with functional MRI highlighted the relationship between the spread of tau pathology and alterations in brain functional connectivity in AD, supporting hypotheses for the trans-neuronal propagation of tau pathology [157]. The spread of tau pathology across synaptic connections, in an activity-dependent manner, could also occur in other age-related neurodegenerative diseases.
The dysfunction of cellular degradation systems, including the autophagy-lysosome and ubiquitin-protease systems [158,159], has been linked with oxidative stress, impaired energy metabolism, synaptic dysfunction [158] and the formation of toxic protein aggregates [160,161,162]. Dysfunction of the glymphatic system and sleep disturbances have also been linked with the accumulation of toxic proteins [163]. Across these pathways, it has been postulated that the removal of toxic abnormal protein aggregates may confer neuroprotection [164,165,166,167]. Both the intrinsic and extrinsic apoptosis signaling cascades have also been implicated to play a role in neuronal death and neurodegeneration. However, the exact mechanism and relationship with upstream pathways remains unclear. For example, in PD, LRRK2 mutations have been linked to mitochondria-dependent intrinsic apoptosis pathways [168], while PINK1 and Parkin mutations show protective effects against stress-induced cytochrome c release [169,170]. Neuroinflammation also plays a role as an extracellular driving factor for the activation of extrinsic apoptosis pathways via oxidative insults or proinflammatory cytokines [72,171,172]. While apoptosis could represent a convergence point for different preceding molecular pathways, therapeutic interventions targeting upstream events, prior to activation of the apoptosis pathways, are likely to be more beneficial. PET imaging tools targeting the degradation and apoptotic pathways are currently lacking. The development of such techniques could help to better elucidate their role within the puzzle of molecular pathways underlying protein aggregation and pathobiology.
6. Synaptic Dysfunction
Synaptic dysfunction and the loss of synaptic density has been suggested as a common key feature across age-related neurodegenerative diseases [173,174,175,176,177,178]. Under physiological conditions, α-synuclein [179], tau and amyloid-β [180] play a role in supporting synaptic function. However, the abnormal accumulation of these proteins [180,181], alongside neuroinflammation [182], mitochondrial dysfunction [183] and energy dysregulation [30], has been linked with pathological events, including the loss of synaptic integrity and plasticity [184], as well as neurotransmitter dysfunction [185,186], across several age-related neurodegenerative diseases [187]. Anatomical and physiological synaptic dysfunctions could reflect a shared mechanism across neurodegenerative diseases; however, the upstream molecular components leading to synaptic dysfunction, as well as the downstream consequences, may vary between diseases, depending on the synaptic population affected.
Until recently, it was only possible to study synaptic density in post-mortem brain tissue [188]. However, the development of novel PET tracers targeting the presynaptic vesicle glycoprotein 2A (SV2A), which is critical for Ca2+ dependent exocytosis [189], paved the way for the in vivo investigation of presynaptic integrity (Figure 2D) [190]. More recently, (11C)UCB-J PET has been employed to study SV2A levels in aging [44], idiopathic PD [58,191,192], PSP and CBS [193], as well as across the cognitive spectrum in AD, from preclinical and MCI to advanced clinical dementia [62,194]. In amyloid-β-positive AD patients, decreased (11C)UCB-J binding was observed in the hippocampus, which correlated with episodic memory [194]. Preclinical studies support the relationship between synaptic loss and cognitive decline prior to neuronal loss [177,188,195]. Preliminary work suggests that the presence of tau and amyloid-β pathology is linked with synaptic dysfunction across the cognitive spectrum in AD [196,197]. In early drug-naïve PD patients, SV2A loss has been observed in the caudate, putamen, thalamus, brainstem and dorsal raphe, as well as cortical regions [58], with SV2A loss also reported in the substantia nigra in treated PD patients [191,192] and additional SV2A loss in the red nucleus and locus coeruleus in moderate to advanced PD patients [191]. Reduced SV2A was associated with global disease burden and motor symptom severity in early drug-naïve PD patients [58]. In PSP patients, the loss of SV2A has recently been reported in the medulla, substantia nigra, pallidum, midbrain, pons and caudate nucleus, with greatest SV2A loss in the medulla, hippocampus, amygdala, caudate, insula and thalamus in CBD patients with amyloid-β-negative (11C)PIB PET scans [193]. The global loss of SV2A in PSP and CBS patients correlated with the total PSP and CBD rating scale as a measure of disease severity and with global cognitive dysfunction [193]. Preliminary work suggests SV2A loss could also be present in PDD and DLB patients [198], as well as in patients with FTD [63]. Work is ongoing to investigate the role of SV2A in ALS, using (11C)UCB-J PET as part of the MIND-MAPS program (https://lp.invicro.com/mind-maps), which could help to provide a comprehensive understanding of the role of SV2A across the spectrum of age-related neurodegenerative diseases. Together, these recent findings provide in vivo evidence to implicate synaptic loss in the pathophysiology of age-related neurodegenerative diseases with potential relevance for clinical phenotypes. Ongoing longitudinal (11C)UCB-J PET studies, such as the TRAID study [199] and the MIND-MAPS program, will help to shed light on the role of synaptic dysfunction on disease progression. The relationship of synaptic dysfunction with protein pathology, as well as mitochondrial dysfunction and neuroinflammation, warrants investigation across age-related neurodegenerative disorders to better understand the interplay between these molecular pathways.
Prior to synaptic loss, synaptic function and plasticity is likely to be significantly disrupted. The accumulation of protein aggregates has been linked with dysfunction of synaptic signaling pathways, such as impairing long-term potentiation (LTP) [200,201] and enhancing long-term depression (LTD [202]), through the dysregulation of metabotropic glutamate receptors (mGluR) and AMPA and NMDA receptors [203,204,205]. The aggregation and deposition of toxic proteins can also alter synaptic neurotransmitter release by impairing the dynamics of synaptic vesicle endocytosis, recycling, mobilization and storage [185,206,207]. Alpha-synuclein, tau and amyloid-β have also been linked with the membrane trafficking and function of several neuron-specific transporters, including dopamine transporter (DAT), serotonin transporters (SERT) and norepinephrine transporter (NET), which play a critical role in regulating neurotransmitter reuptake from the synaptic cleft [208,209,210,211], as well as glutamate receptor subunits, which play a role in modulating synaptic transmission [212,213,214]. Furthermore, genetic mutations, such as LRRK2 mutation in PD, have also been shown to impair the dynamics of synaptic vesicle endocytosis, recycling, mobilization and storage [206,207].
7. Neurotransmitter Dysregulation
PET imaging studies have collectively demonstrated disruption of neurotransmitter systems (Figure 2E), illustrating both overlapping and distinctive features across age-related neurodegenerative diseases [215]. Neurotransmitter systems have traditionally been a key target for pharmacological intervention based on accumulated evidence that the dysregulation of neurotransmission is closely linked with clinical phenotypes. Here, we focused on the main neurotransmitter systems, cholinergic, serotonergic, dopaminergic, noradrenergic, glutamatergic and GABAergic, highlighting key findings from PET imaging studies across age-related neurodegenerative diseases and shedding light on potential future directions. It is important to acknowledge the potential role of other neurotransmitter systems, including the histaminergic, adrenergic, opioid and cannabinoid systems, which are not captured within this review.
The cholinergic system is predominately associated with the development of cognitive impairment in AD, PD, parkinsonian plus and FTD [120,216]. (11C)PMP PET, targeting presynaptic acetylcholinesterase (AChE [217]), has been shown to be reduced in AD patients [218], as well as in PD, PDD and DLB patients [219,220,221]. Furthermore, (18F)2FA PET has revealed hippocampal and cortical loss of the postsynaptic α4β2 nicotinic AChE (nACh) receptor in AD [222] and subcortical and cortical losses in PD patients associated with cognitive decline and depression [223]. Despite the known loss of AChE within the Nucleus Basalis of Meynert in post-mortem FTD tissue [224], no changes in AChE activity were detected in vivo, using (18F)MP4A PET in FTD patients [225]. PET studies in PSP, CBS and MSA patients revealed reduced AChE activity within the pons, thalamus and basal ganglia [225,226]. In PSP and MSA, reduced AChE activity in subcortical regions is greater than reductions observed in PD, showing relevance for the development of gait disturbance [226]. Overlap of the cholinergic hypothesis, reflecting neuronal loss in the Nucleus Basalis of Meynert, associated with cortical cholinergic deficits and cognitive impairments, has also been highlighted across AD, PDD and PD [227,228,229,230]. Therefore, while cholinergic dysfunction is likely present across several age-related neurodegenerative diseases, the degree of deficits, as well as the temporal and spatial presentations, is likely different. Future PET imaging of the vesicular acetylcholine transporters, using (18F)FEOBV [231], and of the α7-nACh receptors using the novel tracer (18F)ASEM [232,233], could provide further insights into the integrity of cholinergic nerve terminals in vivo.
Looking at the serotonergic system, (11C)DASB PET has most widely been employed in PD, showing serotonergic pathology starting in premotor stages before the development of overt motor symptoms [234,235] through to advanced disease stages [236]. Serotonergic dysfunction in PD has been linked with the development of nonmotor clinical phenotypes, including depression [237], sleep diseases [238], fatigue [239], weight changes [240], apathy [241] and visual hallucinations [242], in addition to the development of motor phenotypes of tremor [243], levodopa-induced dyskinesias [244] and graft-induced dyskinesias [245]. Albeit to a lesser extent, the serotonergic system has also been implicated in AD, ALS and FTD [246]. (18F)MPPF PET studies showing reduced levels of serotonin type 1A (5-HT1A) receptors in the hippocampus of AD patients [247]. The development of PET tracers targeting the 5-HT6 receptor, such as (18F)2FNQ1P [248,249], could play an important role to further explore the relationship of this serotonin receptor subtype with dementia. In FTD, the loss of 5-HT2A receptors, measured using (11C)MDL100907 PET, has been reported to underlie behavior diseases [250]. A (11C)WAY100635 PET study also reported decreased 5-HT1A receptor binding in ALS patients within the frontotemporal and cingulate regions [251,252]. Therefore, serotonergic dysfunction across different diseases could involve different serotonin receptor subtypes. (11C)Cimbi-36 PET, a 5-HT2A receptor agonist, could offer a valuable tool to investigate changes in serotonin synaptic levels and, indirectly, to measure serotonin release [253,254]. The serotonergic system also interacts with and regulates multiple other neurotransmitter systems, including the dopaminergic, glutamatergic, GABAergic and noradrenergic systems [255]. Given the complexity of the serotonergic system, its central modulatory influences and links with various clinical phenotypes, it could present a principal orchestrator of disease-related pathology and may, perhaps, hold the potential to significantly affect the disease course.
The dopaminergic system underpins the development of primary motor and extrapyramidal phenotypes in PD, parkinsonian plus, HD and FTD but has also been linked with behavioral and cognitive phenotypes in PD, parkinsonian plus, FTD [215,256] and, to some extent, AD [257,258] and ALS [259]. Several PET tracers are available to measure postsynaptic dopamine receptors, such as (11C)Raclopride, (11C)PHNO and (18F)Fallypride, as well as presynaptic dopamine integrity assessing DAT with (11C)PE2I, dopamine storage with (18F)DOPA and vesicle monoamine transporter type 1 with (11C)DTBZ [256,260]. While presynaptic PET markers show some variations, the overarching findings indicate that presynaptic dopaminergic integrity is impaired across PD, PSP, CBS and FTD, associated mainly with rigidity and bradykinesia motor symptoms [215,261]. Across different familial forms of PD, differing patterns of striatal presynaptic dopaminergic loss have been reported, which could reflect the differential influence of specific genetic mutations on molecular pathways towards the etiopathogenesis and progression of PD [262,263]. D2 receptor binding is relatively preserved in PD, while D2 receptor binding is reduced in PSP, MSA and HD; in the latter, decreased D2 receptor binding can be observed during presymptomatic disease stages [264]. A combined (18F)DOPA and (11C)MP4A PET study illustrated that PDD and DLB share similar dopaminergic and cholinergic deficit profiles [221]. Presynaptic and postsynaptic dopaminergic dysfunctions have also been linked with cognitive and executive dysfunction in PD [265,266,267,268,269,270,271]. The presence and potential role of dopaminergic dysfunction remains unclear in AD, with a recent report suggesting that dopaminergic system could be linked with the pathophysiology of AD [258], whilst another showed no changes in DAT uptake in early-onset AD patients [272]. Decreased striatal DAT binding was also reported in ALS patients [259], although the role of dopaminergic dysfunction in ALS still remains to be elucidated.
The noradrenergic system has been linked with the regulation of several autonomic functions and behavioral and cognitive phenotypes, such as attention, wakefulness, decision making, memory and depression [273,274,275,276]. While there is growing evidence to implicate noradrenergic dysfunction across age-related neurodegenerative diseases [273,277], in vivo studies are generally lacking due to the absence of suitable radioligands. (11C)MRB PET, a selective ligand for NET, has been employed to study noradrenergic synaptic terminals in PD [278,279,280]. The PET radioligand (11C)RTI-32, a marker of both DAT and NET, has also been employed in PD, with evidence suggesting that the loss of dopaminergic and noradrenergic innervation within the limbic system may play a role in the development of depression and anxiety in the PD disease course [281]. Future studies are warranted, alongside the development of novel PET radioligands, to deepen our understanding of the role of the noradrenergic system across the spectrum of different age-related neurodegenerative diseases and the direct relevance for specific clinical phenotypes.
Glutamatergic neurotransmission is also known to play a central role in supporting higher cognitive function. Excessive glutamate neurotransmission can promote excitotoxic neuronal death contributing to the glutamate hypothesis in AD and FTD [282,283,284]. Recently, (18F)FPEB and (11C)ABP688 PET studies demonstrated a reduction of hippocampal mGluR5 in early AD, which was associated with lower episodic memory scores and reduced global cognitive function [285,286]. While the relevance of hippocampal mGluR5 loss in AD needs to be fully elucidated, taken together with findings of decreased hippocampal (11C)UCB-J binding in AD [194], the loss of mGluR5 could reflect nonspecific synaptic loss. Conversely, the specific synaptotoxicity of amyloid-β at mGluR sites could influence the spatial distribution of mGlu5R loss [287] and could be related to excitotoxin-induced neurodegeneration [288]. Reductions of (11C)ABP688 PET uptake have also been reported in patients with the behavior variant FTD (bvFTD) [289]. PET tracers targeting the other metabotropic glutamate receptor subtypes have also been developed, such as the (11C)ITMM and (18F)FIMX radioligands, which are specific for mGluR1 [290,291]. Given the reported loss of mGluR1 expression in DLB [292], it could be of interest to investigate the role of mGluR1 across several age-related neurodegenerative diseases utilizing these PET radioligands.
The GABAergic system represents the main inhibitory neurotransmitter system within the brain and plays an important role in regulating oscillatory dynamics for cognitive control and work memory function [293,294]. Impaired GABAergic neurotransmission has been linked with HD, FTD, ALS and parkinsonian plus diseases, including PSP. Decreased (11C)Flumazenil PET binding, a marker of GABA-A receptor, has been reported in AD [295], PSP [296] and manifest HD [297,298] and linked with motor and extra-motor cortical changes in sporadic ALS [299,300]. The findings that reduced hippocampal (11C)Flumazenil PET-binding correlates with memory performance in early AD are in accordance with post-mortem evidence of reduced hippocampal GABA-A messenger RNA expression [295].
8. Dysfunction of the Glymphatic System
The glymphatic system is proposed to remove waste products through the cerebrospinal fluid (CSF) to interstitial fluid (ISF) exchange, along the perivascular pathway, with the help of aquaporin-4 water channels, which are polarized in astrocytic end feet towards the capillary vessel walls [163,301,302]. Glymphatic clearance is most active during sleep when aquaporin-4 water channels expand, allowing the removal of metabolic and protein waste products that accumulate during wakefulness [303]. The potential importance of the glymphatic system in the clearance of waste products, including pathogenic protein aggregates, has sparked growing interest to unravel the role of the glymphatic system and sleep dysfunction in the development and progression of age-related neurodegenerative diseases. It can be hypothesized that dysfunction of the glymphatic system, potentially linked with sleep disturbances, may be a common contributing pathogenic factor to several age-related neurodegenerative diseases and, as such, a potential modulable target for therapeutic intervention. The exact relationship between sleep problems, glymphatic dysregulation and protein accumulation in late-onset neurodegenerative diseases remains to be further elucidated.
Dysfunction of the glymphatic system, in late life, has been shown to contribute to the accumulation of amyloid-β [304,305]. Aquaporin-4 knockout mice showed worse cognitive performance and increased accumulation of amyloid-β in the parenchyma and perivascular space compared to wild-type mice [306]. A post-mortem study revealed reduced perivascular end-feet localization of aquaporin-4 in the brains of AD patients [307]. Therefore, redistribution of aquaporin-4 in AD could lead to the dysfunction of glymphatic clearance, resulting in amyloid-β accumulation. Alternatively, the redistribution of aquaporin-4 could be part of the mechanistic actions underlying glymphatic dysfunction. Recently, genetic variants of aquaporin-4 have been associated with sleep disturbances, amyloid-β burden and clinical conversion from MCI to AD [308,309,310]. While the precise role and interactions of aquaporin-4 with the glymphatic system have not been fully elucidated, these findings further support the hypothesis that dysfunction of the glymphatic system could play a pivotal role in neurodegeneration and disease pathogenesis. The PET radioligand (11C)TGN-020 has been developed for the quantification of aquaporin-4 [311]. In humans, the distribution of (11C)TGN-020 within the brain is consistent with known distributions of aquaporin-4—namely, subpial and perivascular end-feet of astrocytes and choroid plexus [311]. Therefore, (11C)TGN-020 PET could have useful applications to help unravel the molecular mechanism by which aquaporin-4 interacts with the glymphatic system and the relationship with disease pathology in age-related neurodegenerative diseases.
Glymphatic MRI, with intrathecal injections of MRI-based contrasts acting as a CSF tracer, has been developed to quantify CSF-ISF exchange in vivo [312,313]. Recently, delayed clearance of the CSF tracer in the entorhinal cortex was observed in cognitively impaired patients with idiopathic normal pressure hydrocephalus [314,315], a potentially treatable form of cognitive impairment, in late life [316], which may exhibit AD-like pathological features, including the deposition of amyloid-β and tau [317]. The in vivo findings that patients with cognitive deficits display delayed clearance of the CSF tracer within regions involved with cognitive function further supports the hypothesis that dysfunction of the glymphatic system may contribute to the development of dementia and merits further investigations in the preclinical disease stages. The development of a PET radioligand to assess glymphatic function at a molecular level could help to better delineate the role of glymphatic dysfunction in disease etiopathogenesis and pathophysiology.
The development of novel glymphatic system MRI methodologies, such as 3D phase-contrast MRI [318,319,320,321,322,323,324,325,326], ultra-fast encephalography MRI [327], near-infrared spectroscopy and ultra-fast functional MRI [328], have gained growing interest to better understand fluid dynamics within the brain and to study the role of neuro-hydrodynamics with respect to the glymphatic system. Arterial spin labeling and diffusion tensor imaging techniques are also being employed to assess CBF crossing the blood-brain interface [329] and the movement of fluid in the perivascular space [330,331], respectively. Seven tesla MRI techniques have been proposed as the optimal imaging modality to successfully measure the perivascular space [332,333,334]. The topographical distribution of high-grade MRI-visible perivascular spaces in patients with vascular dementia, cerebral amyloid angiopathy and AD may be overlapping but may also occur in distinct brain regions [335,336,337]. Furthermore, dilated perivascular spaces in the basal ganglia have been linked with high total tau levels in the CSF, indicating neurodegeneration [335]. Interestingly, it has been reported that the severity of MRI-visible perivascular spaces in the basal ganglia are associated with clinically diagnosed subcortical vascular cognitive impairment and negatively predicted AD, whilst the severity of such lesions in the centrum semi-ovale is associated with clinically diagnosed AD [334]. Such studies provide further in vivo evidence on the role of glymphatic dysfunction in different late-onset dementia types. Mathematical models of the glymphatic system are also being developed to better understand the glymphatic system dynamics and pathways under physiological and pathological conditions, as well as to provide quantitative maps for understanding disease pathophysiology and monitoring disease progression [338,339,340,341,342,343]. The continuous development of noninvasive imaging techniques and computational models will allow for the wider application of future in vivo studies investigating the role of the glymphatic system across the spectrum of age-related neurodegenerative diseases.
Preclinical animal models have shown that the clearance of amyloid-β is dysregulated under conditions of sleep deprivation and with advanced aging [344,345,346,347]. Preliminary in vivo studies support the role of sleep in perivascular clearance [348]. In healthy humans, the amyloid-β burden is increased after one night of sleep deprivation [349]. An (18F)FDG PET study revealed a reorganization of regional cerebral metabolic activity following sleep deprivation in healthy individuals, which was associated with the decline in cognitive performance observed after sleep deprivation [350]. PET studies using (11C)PiB to measure the brain amyloid-β load have demonstrated that adults who report less adequate sleep, more sleep problems and greater somnolence have a greater amyloid-β burden in AD-sensitive brain regions, independently of the APOE-ε4 genotype [351,352]. Based on the above data, it could be hypothesized that, in older adults, sleep disturbances may lead to an increasing dysfunction of the glymphatic system, which could subsequently contribute to increasing the amyloid-β and tau loads. However, the temporal sequence of sleep problems and disease onset, i.e., whether they represent a contributing pathogenic factor (in the preclinical AD stages) or an early clinical manifestation of MCI due to AD, still remains to be determined [347,353]. PET studies performed in healthy volunteers have also implicated changes within the dopaminergic and serotonergic systems related to sleep deprivation, including downregulation of the D2/D3 receptors in the ventral striatum [354,355,356], with no changes in dopamine release [354] and an upregulation of the 5-HT2A receptors in frontal and parietal cortices [357]. Together, these findings suggest a potential interplay between sleep, glymphatic dysfunction and disease pathology, which merits future investigations across several neurodegenerative diseases associated with aging.
9. PET Imaging and Personalized Precision Medicine in Advancing the Precision Medicine Icon to Aid Sub-Phenotyping
Challenging the traditional clinicopathological nosology of individual age-related neurodegenerative diseases may open avenues for dissecting disease entities, such as AD or PD, and unraveling their potential heterogeneity into subtypes through in vivo multifaceted biomarker studies. Each with a distinct biomarker-based signature profile, reflecting its distinct pathogenic pathway(s). This approach is not dissimilar to the stratification paradigm, which has, recently, proven its value in several cancer types. Significant recent advances in cancer biology and a biomarker-based subclassification allowed for targeted precision pharmacological interventions, showing significant therapeutic benefit in evidence-based stratified patients, whereas they had previously failed to meet the desired effects [358]. Genetic-based risk prediction models, alongside imaging biomarkers and known risk factors involving vascular and metabolic comorbidities and lifestyle factors, are currently being evaluated for stratifying aging individuals into AD risk groups [359,360,361,362,363,364,365,366,367,368]. Furthermore, disease-specific network patterns, identified using PET and functional MRI techniques, could prove beneficial to unravel network alterations across diseases and to determine the effectiveness of therapies aiming to modulate underlying disease pathways [156,369]. The in vivo investigation of epigenetics, from the preclinical stages and during disease progression, could unlock new insights into gene regulatory processes, disease pathophysiology and novel targets for therapeutic intervention [370]. Imaging epigenetics may now be possible with the recent development of the (11C)Martinostat PET radioligand to measure class I histone deacetylase (HDAC) enzyme density [371].
While PET imaging can be complex, expensive and its wider application may be limited due to infrastructural and logistical constraints, PET imaging remains the only reliable noninvasive in vivo solution to quantify molecular targets and pathologies in patients across the disease life course. The continued development and wider application could help to transfer promising PET techniques from a research to a clinical setting. There is a need for more longitudinal studies to answer a number of key neurobiological questions and to determine the use of PET radioligands as reliable biomarkers to track disease progression. Furthermore, future work should focus on understanding the relationship between PET biomarkers and early signs and symptoms of age-related neurodegenerative diseases. Capitalizing on fully penetrant genetic mutations could allow for the investigation and characterization of early disease pathology in preclinical stages and, accompanied with longitudinal follow-ups, could help to shed further light on the relationship between these PET markers and early disease symptomatology. Recent methodological advances to measure amyloid-β and tau loads from PET imaging, with higher sensitivity and lower variability, could further improve the accuracy of imaging phenotypic stratification and increased power to detect meaningful outcomes and biological effects in future clinical trials [372,373]. Novel tools such as AmyloidIQ and TauIQ may add future value to the recent AT(N) classification of AD [10,11]. The scientific community should strive to deepen and enhance these approaches by capitalizing on the next-generation PET radioligands and novel PET biomarkers (Table 1), combined with advanced MRI and computational methodologies. Advances in the next generation of PET radioligands for neuroinflammation, alongside the development of novel radioligands targeting mitochondrial functions, could help to unravel the interplay between glia-related neuroinflammation, abnormal protein aggregation and mitochondrial dysregulation. In the case of neuroinflammation, there is a need to better understand how TSPO changes relate to underlying pathology and to further explore novel molecular targets beyond TSPO, such as cyclooxygenase, purinergic receptors and astrocytes. The development of a PET radioligand specific for α-synuclein, Huntingtin and TDP-43 would be ground-breaking and result in significant advances within the field, including aiding diagnostics; tracking disease progression and the ability to monitor and assess novel drugs aiming to reduce α-synuclein, Huntingtin or TDP-43 levels. Furthermore, the development of such PET radioligands could aid the stratification of patients into future clinical trials and, potentially, help to drive the treatment landscape towards the precision medicine paradigm.
10. Conclusions
The scientific community is slowly moving away from the traditional clinicopathological disease models towards a precision medicine paradigm, which is still lacking in age-related neurodegenerative diseases. Based on the available understanding of interlinked genomic, biological and clinical pathways across diseases, it is likely that a dynamic multifaceted biomarker approach is required to disentangle mechanisms of disease pathogenesis and clinical trajectories. This might include markers of pathology spanning from genetic, epigenetic, proteomic, metabolomic and transcriptomic, clinical and digital and molecular PET and MRI imaging to biological CSF and blood-based biomarkers. Unraveling disease-specific network changes underpinned by genetic and pathological patterns could aid the advancement of personalized precision medicine by providing novel diagnostic tools and stratification approaches, as well as in identifying new therapeutic targets. Molecular PET imaging will likely continue to offer an invaluable tool, reflecting molecular pathology, contributing towards unlocking disease mechanisms and identifying biomarkers throughout the disease course. It is hoped that advances in the development of novel PET radioligands (Table 1) and their applications in age-related neurodegenerative diseases, as well as in healthy aging, will help to drive advancements in sub-phenotyping to aid precision medicine-based approaches in diagnosis, prevention and clinical management.
Author Contributions
Conceptualization and design, L.T.M. and H.W.; writing—original draft preparation, H.W. and writing—review and editing, M.P., E.A.R. and L.T.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
E.A.R. is an employee of Invicro LLC. The authors declare no conflict of interest.
References
- United Nations, Department of Economic and Social Affairs. Population Division. World Population Ageing; United Nations: New York, NY, USA, 2013.
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.M.; Devenney, E.M.; Irish, M.; Ittner, A.; Naismith, S.; Ittner, L.M.; Rohrer, J.D.; Halliday, G.M.; Eisen, A.; Hodges, J.R.; et al. Neuronal network disintegration: Common pathways linking neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1234–1241. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.D.; Rohrer, J.D.; Schott, J.M.; Fox, N.C.; Hardy, J.; Rossor, M.N. Molecular nexopathies: A new paradigm of neurodegenerative disease. Trends Neurosci. 2013, 36, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Arneson, D.; Zhang, Y.; Yang, X.; Narayanan, M. Shared mechanisms among neurodegenerative diseases: From genetic factors to gene networks. J. Genet. 2018, 97, 795–806. [Google Scholar] [CrossRef]
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Dissecting the Molecular Mechanisms of Neurodegenerative Diseases through Network Biology. Front. Aging Neurosci. 2017, 9, 166. [Google Scholar] [CrossRef] [Green Version]
- Pievani, M.; de Haan, W.; Wu, T.; Seeley, W.W.; Frisoni, G.B. Functional network disruption in the degenerative dementias. Lancet Neurol. 2011, 10, 829–843. [Google Scholar] [CrossRef] [Green Version]
- Eisen, A.; Turner, M.R. Does variation in neurodegenerative disease susceptibility and phenotype reflect cerebral differences at the network level? Amyotroph Lateral Scler. Front. Degener. 2013, 14, 487–493. [Google Scholar] [CrossRef]
- Chhatwal, J.P.; Schultz, A.P.; Johnson, K.A.; Hedden, T.; Jaimes, S.; Benzinger, T.L.S.; Jack, C., Jr.; Ances, B.M.; Ringman, J.M.; Marcus, D.S.; et al. Preferential degradation of cognitive networks differentiates Alzheimer’s disease from ageing. Brain 2018, 141, 1486–1500. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- McRae-McKee, K.; Udeh-Momoh, C.T.; Price, G.; Bajaj, S.; de Jager, C.A.; Scott, D.; Hadjichrysanthou, C.; McNaughton, E.; Bracoud, L.; Ahmadi-Abhari, S.; et al. Perspective: Clinical relevance of the dichotomous classification of Alzheimer’s disease biomarkers: Should there be a “gray zone”? Alzheimers Dement. 2019, 15, 1348–1356. [Google Scholar] [CrossRef]
- Sturchio, A.; Marsili, L.; Mahajan, A.; Grimberg, M.B.; Kauffman, M.A.; Espay, A.J. How have advances in genetic technology modified movement disorder nosology? Eur. J. Neurol. 2020, 27, 1461–1470. [Google Scholar] [CrossRef]
- Burns, T.C.; Verfaillie, C.M. From mice to mind: Strategies and progress in translating neuroregeneration. Eur. J. Pharm. 2015, 759, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef]
- Burns, T.C.; Li, M.D.; Mehta, S.; Awad, A.J.; Morgan, A.A. Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: A systematic bioinformatics-based critique of preclinical models. Eur. J. Pharm. 2015, 759, 101–117. [Google Scholar] [CrossRef]
- Johnson, I.P. Age-related neurodegenerative disease research needs aging models. Front. Aging Neurosci. 2015, 7, 168. [Google Scholar] [CrossRef]
- Kobylecki, C.; Jones, M.; Thompson, J.C.; Richardson, A.M.; Neary, D.; Mann, D.M.; Snowden, J.S.; Gerhard, A. Cognitive-behavioural features of progressive supranuclear palsy syndrome overlap with frontotemporal dementia. J. Neurol. 2015, 262, 916–922. [Google Scholar] [CrossRef]
- Mioshi, E.; Caga, J.; Lillo, P.; Hsieh, S.; Ramsey, E.; Devenney, E.; Hornberger, M.; Hodges, J.R.; Kiernan, M.C. Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology 2014, 82, 149–155. [Google Scholar] [CrossRef]
- Borroni, B.; Alberici, A.; Agosti, C.; Cosseddu, M.; Padovani, A. Pattern of behavioral disturbances in corticobasal degeneration syndrome and progressive supranuclear palsy. Int. Psychogeriatr. 2009, 21, 463–468. [Google Scholar] [CrossRef]
- Li, M.D.; Burns, T.C.; Morgan, A.A.; Khatri, P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol. Commun. 2014, 2, 93. [Google Scholar] [CrossRef]
- Millan, M.J. An epigenetic framework for neurodevelopmental disorders: From pathogenesis to potential therapy. Neuropharmacology 2013, 68, 2–82. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharm. 2019, 10, 902. [Google Scholar] [CrossRef]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharm. Exp. 2012, 342, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S499–S512. [Google Scholar] [CrossRef] [Green Version]
- Roses, A.; Sundseth, S.; Saunders, A.; Gottschalk, W.; Burns, D.; Lutz, M. Understanding the genetics of APOE and TOMM40 and role of mitochondrial structure and function in clinical pharmacology of Alzheimer’s disease. Alzheimers Dement. 2016, 12, 687–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokopenko, I.; Miyakawa, G.; Zheng, B.; Heikkinen, J.; Petrova Quayle, D.; Udeh-Momoh, C.; Claringbould, A.; Neumann, J.; Haytural, H.; Kaakinen, M.A.; et al. Alzheimer’s disease pathology explains association between dementia with Lewy bodies and APOE-epsilon4/TOMM40 long poly-T repeat allele variants. Alzheimers Dement. (N. Y.) 2019, 5, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Chiba-Falek, O.; Gottschalk, W.K.; Lutz, M.W. The effects of the TOMM40 poly-T alleles on Alzheimer’s disease phenotypes. Alzheimers Dement. 2018, 14, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschalk, W.K.; Lutz, M.W.; He, Y.T.; Saunders, A.M.; Burns, D.K.; Roses, A.D.; Chiba-Falek, O. The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging. J. Parkinsons Dis. Alzheimers Dis. 2014, 1. [Google Scholar] [CrossRef]
- Ridge, P.G.; Kauwe, J.S.K. Mitochondria and Alzheimer’s Disease: The Role of Mitochondrial Genetic Variation. Curr. Genet. Med. Rep. 2018, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Martinez, A.; Blanco, R.; Dalfo, E.; Carmona, M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: Preclinical Parkinson disease. J. Neural Transm. 2011, 118, 821–839. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Hunn, B.H.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 2015, 38, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Zaltieri, M.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.; Bellucci, A. Mitochondrial Dysfunction and alpha-Synuclein Synaptic Pathology in Parkinson’s Disease: Who’s on First? Parkinsons Dis. 2015, 2015, 108029. [Google Scholar] [CrossRef]
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A.; et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636. [Google Scholar] [CrossRef]
- Mansur, A.; Rabiner, E.A.; Comley, R.A.; Lewis, Y.; Middleton, L.T.; Huiban, M.; Passchier, J.; Tsukada, H.; Gunn, R.N.; Consortium, M.-M. Characterization of 3 PET Tracers for Quantification of Mitochondrial and Synaptic Function in Healthy Human Brain: (18)F-BCPP-EF, (11)C-SA-4503, and (11)C-UCB-J. J. Nucl. Med. 2020, 61, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Mansur, A.; Rabiner, E.A.; Tsukada, H.; Comley, R.A.; Lewis, Y.; Huiban, M.; Passchier, J.; Gunn, R.N. Test-retest variability and reference region-based quantification of (18)F-BCPP-EF for imaging mitochondrial complex I in the human brain. J. Cereb. Blood Flow Metab. 2020. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Francardo, V.; Bez, F.; Wieloch, T.; Nissbrandt, H.; Ruscher, K.; Cenci, M.A. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 2014, 137, 1998–2014. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Y.; Pokrass, M.J.; Klauer, N.R.; De Credico, N.E.; Su, T.P. Sigma-1 receptor chaperones in neurodegenerative and psychiatric disorders. Expert Opin. Ther. Targets 2014, 18, 1461–1476. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; De Rasmo, D. Complex I deficiencies in neurological disorders. Trends Mol. Med. 2013, 19, 61–69. [Google Scholar] [CrossRef]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Guo, L.W.; Epstein, M.L.; Ruoho, A.E. Role of the Sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS). J. Pharm. Sci. 2015, 127, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Jansen, K.L.; Faull, R.L.; Storey, P.; Leslie, R.A. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer’s disease correlates with CA1 pyramidal cell loss. Brain Res. 1993, 623, 299–302. [Google Scholar] [CrossRef]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–152. [Google Scholar] [CrossRef] [Green Version]
- Flones, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; et al. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018, 135, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Wilson, H.; Pagano, G.; de Natale, E.R.; Mansur, A.; Caminiti, S.P.; Polychronis, S.; Middleton, L.T.; Price, G.; Schmidt, K.F.; Gunn, R.N.; et al. Mitochondrial Complex 1, Sigma 1, and Synaptic Vesicle 2A in Early Drug-Naive Parkinson’s Disease. Mov. Disord. 2020, 35, 1416–1427. [Google Scholar] [CrossRef]
- Toyohara, J.; Sakata, M.; Ishiwata, K. Imaging of sigma1 receptors in the human brain using PET and [11C]SA4503. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 190–196. [Google Scholar] [CrossRef]
- Mishina, M.; Ishiwata, K.; Ishii, K.; Kitamura, S.; Kimura, Y.; Kawamura, K.; Oda, K.; Sasaki, T.; Sakayori, O.; Hamamoto, M.; et al. Function of sigma1 receptors in Parkinson’s disease. Acta Neurol. Scand. 2005, 112, 103–107. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ohba, H.; Nishiyama, S.; Kakiuchi, T.; Tsukada, H. Effect of MPTP on Serotonergic Neuronal Systems and Mitochondrial Complex I Activity in the Living Brain: A PET Study on Conscious Rhesus Monkeys. J. Nucl. Med. 2017, 58, 1111–1116. [Google Scholar] [CrossRef]
- Venkataraman, A.; Mansur, A.; Lewis, Y.; Kocagoncu, E.; Lingford-Hughes, A.; Huiban, M.; Passchier, J.; Rowe, J.; Tsukada, H.; Brooks, D.; et al. Evaluation of mitochondrial and synaptic and synaptic function in Alzheimer’s disease: A [18F]BCPP-EF, [11C]SA4503 and [11C]UCB-J PET study. In Proceedings of the 29 International Symposium on Cerebral Blood Flow, Metabolism and Function, Yokohama, Japan, 4–7 July 2019; Volume 39, pp. 121–122. [Google Scholar]
- Clarke, M.; Mansur, A.; Passchier, J.; Lewis, Y.; Evans, K.; Chen, L.; Schwarz, A.; Takano, A.; Gunn, R.; Cash, D.; et al. Imaging synaptic and mitochondrial function in frontotemporal dementia using [11C]UCB-J, [18F]BCPP-EF and [11C]SA4503 PET. In Proceedings of the Human Amyloid Imaging, Miami, FL, USA, 15–17 January 2020. [Google Scholar]
- Rabiner, E.; Mansur, A.; Venkataraman, A.; Price, G.; Wilson, H.; Pagano, G.; Clarke, M.; Lewis, Y.; Matthews, P.M.; Rowe, J.B.; et al. MIND MAPS: Assessment of the mitochondrial—Endoplasmic reticulum—Synaptic axis in neurodegeneration by [18F]BCPP-EF, [11C]SA4503 and [11C]UCB-J PET imaging. In Proceedings of the Human Amyloid Imaging, Miami, FL, USA, 15–17 January 2020. [Google Scholar]
- Terada, T.; Therriault, J.; Su, P.K.M.; Savard, M.; Ouchi, Y.; Rosa-Neto, P. In vivo association of mitochondrial dysfunction with tau pathology in early Alzheimer’s disease. In Proceedings of the Human Amyloid Imaging, Miami, FL, USA, 15–17 January 2020. [Google Scholar]
- Mosley, R.L.; Hutter-Saunders, J.A.; Stone, D.K.; Gendelman, H.E. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009381. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Rowe, D.B.; Carew-Jones, F.M.; Hayes, M.; Chan, D.K.; Blackie, J.; Corbett, A.; Joffe, R.; Fung, V.S.; Morris, J.; et al. Anti-melanin antibodies are increased in sera in Parkinson’s disease. Exp. Neurol. 2009, 217, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Stone, D.K.; Hutter, J.A.; Benner, E.J.; Mosley, R.L.; Gendelman, H.E. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J. Immunol. 2010, 184, 2261–2271. [Google Scholar] [CrossRef] [Green Version]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S.M.; Cristovao, A.C.; Campos, F.L.; Fonseca, C.P.; Baltazar, G. Astrocyte-derived GDNF is a potent inhibitor of microglial activation. Neurobiol. Dis. 2012, 47, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 69, 337–342. [Google Scholar] [CrossRef]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, C.M.; Annese, V.; Gomez, A.; Ros-Bernal, F.; Aguado-Llera, D.; Martinez-Pagan, M.E.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2012, 3, e379. [Google Scholar] [CrossRef] [Green Version]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [Green Version]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Rodrigues, E.; Jung, J.; Luzentales-Simpson, M.; Enterina, J.R.; Galleguillos, D.; St Laurent, C.D.; Nakhaei-Nejad, M.; Fuchsberger, F.F.; Streith, L.; et al. Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun. Biol. 2019, 2, 450. [Google Scholar] [CrossRef] [Green Version]
- Ikawa, M.; Lohith, T.G.; Shrestha, S.; Telu, S.; Zoghbi, S.S.; Castellano, S.; Taliani, S.; Da Settimo, F.; Fujita, M.; Pike, V.W.; et al. 11C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J. Nucl. Med. 2017, 58, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Kobayashi, M.; Ikawa, M.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Zoghbi, S.S.; Haskali, M.B.; Telu, S.; Pike, V.W.; et al. Comparison of four (11)C-labeled PET ligands to quantify translocator protein 18 kDa (TSPO) in human brain: (R)-PK11195, PBR28, DPA-713, and ER176-based on recent publications that measured specific-to-non-displaceable ratios. EJNMMI Res. 2017, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Su, P.; Piccini, P. Imaging of microglia in patients with neurodegenerative disorders. Front. Pharm. 2012, 3, 96. [Google Scholar] [CrossRef] [Green Version]
- Ghadery, C.; Koshimori, Y.; Coakeley, S.; Harris, M.; Rusjan, P.; Kim, J.; Houle, S.; Strafella, A.P. Microglial activation in Parkinson’s disease using [(18)F]-FEPPA. J. Neuroinflammation 2017, 14, 8. [Google Scholar] [CrossRef] [Green Version]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: A positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef]
- Kang, Y.; Mozley, P.D.; Verma, A.; Schlyer, D.; Henchcliffe, C.; Gauthier, S.A.; Chiao, P.C.; He, B.; Nikolopoulou, A.; Logan, J.; et al. Noninvasive PK11195-PET Image Analysis Techniques Can Detect Abnormal Cerebral Microglial Activation in Parkinson’s Disease. J. Neuroimaging 2018, 28, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Edison, P.; Ahmed, I.; Fan, Z.; Hinz, R.; Gelosa, G.; Ray Chaudhuri, K.; Walker, Z.; Turkheimer, F.E.; Brooks, D.J. Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology 2013, 38, 938–949. [Google Scholar] [CrossRef]
- Gerhard, A.; Banati, R.B.; Goerres, G.B.; Cagnin, A.; Myers, R.; Gunn, R.N.; Turkheimer, F.; Good, C.D.; Mathias, C.J.; Quinn, N.; et al. [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 2003, 61, 686–689. [Google Scholar] [CrossRef]
- Gerhard, A.; Trender-Gerhard, I.; Turkheimer, F.; Quinn, N.P.; Bhatia, K.P.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov. Disord. 2006, 21, 89–93. [Google Scholar] [CrossRef]
- Gerhard, A.; Watts, J.; Trender-Gerhard, I.; Turkheimer, F.; Banati, R.B.; Bhatia, K.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov. Disord. 2004, 19, 1221–1226. [Google Scholar] [CrossRef]
- Lois, C.; Gonzalez, I.; Izquierdo-Garcia, D.; Zurcher, N.R.; Wilkens, P.; Loggia, M.L.; Hooker, J.M.; Rosas, H.D. Neuroinflammation in Huntington’s Disease: New Insights with (11)C-PBR28 PET/MRI. ACS Chem. Neurosci. 2018, 9, 2563–2571. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. (11)C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. NeuroBiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Femminella, G.D.; Dani, M.; Wood, M.; Fan, Z.; Calsolaro, V.; Atkinson, R.; Edginton, T.; Hinz, R.; Brooks, D.J.; Edison, P. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, e1331–e1343. [Google Scholar] [CrossRef] [Green Version]
- Cagnin, A.; Rossor, M.; Sampson, E.L.; Mackinnon, T.; Banati, R.B. In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 2004, 56, 894–897. [Google Scholar] [CrossRef]
- Miyoshi, M.; Shinotoh, H.; Wszolek, Z.K.; Strongosky, A.J.; Shimada, H.; Arakawa, R.; Higuchi, M.; Ikoma, Y.; Yasuno, F.; Fukushi, K.; et al. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: A PET and MRI study. Parkinsonism Relat. Disord. 2010, 16, 404–408. [Google Scholar] [CrossRef]
- Alshikho, M.J.; Zurcher, N.R.; Loggia, M.L.; Cernasov, P.; Reynolds, B.; Pijanowski, O.; Chonde, D.B.; Izquierdo Garcia, D.; Mainero, C.; Catana, C.; et al. Integrated magnetic resonance imaging and [(11) C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 1186–1197. [Google Scholar] [CrossRef]
- Van Weehaeghe, D.; Van Schoor, E.; De Vocht, J.; Koole, M.; Attili, B.; Celen, S.; Declercq, L.; Thal, D.R.; Van Damme, P.; Bormans, G.; et al. TSPO Versus P2X7 as a Target for Neuroinflammation: An In Vitro and In Vivo Study. J. Nucl. Med. 2020, 61, 604–607. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Aman, Y.; Ahmed, I.; Chetelat, G.; Landeau, B.; Ray Chaudhuri, K.; Brooks, D.J.; Edison, P. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement. 2015, 11, 608–621. [Google Scholar] [CrossRef]
- Chauveau, F.; Van Camp, N.; Dolle, F.; Kuhnast, B.; Hinnen, F.; Damont, A.; Boutin, H.; James, M.; Kassiou, M.; Tavitian, B. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J. Nucl. Med. 2009, 50, 468–476. [Google Scholar] [CrossRef] [Green Version]
- McCluskey, S.P.; Plisson, C.; Rabiner, E.A.; Howes, O. Advances in CNS PET: The state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 451–489. [Google Scholar] [CrossRef] [Green Version]
- Narayanaswami, V.; Dahl, K.; Bernard-Gauthier, V.; Josephson, L.; Cumming, P.; Vasdev, N. Emerging PET Radiotracers and Targets for Imaging of Neuroinflammation in Neurodegenerative Diseases: Outlook Beyond TSPO. Mol. Imaging 2018, 17, 1536012118792317. [Google Scholar] [CrossRef] [Green Version]
- Tyacke, R.J.; Myers, J.F.M.; Venkataraman, A.; Mick, I.; Turton, S.; Passchier, J.; Husbands, S.M.; Rabiner, E.I.A.; Gunn, R.N.; Murphy, P.S.; et al. Evaluation of (11)C-BU99008, a positron emission tomography ligand for the Imidazoline2 binding site in human brain. J. Nucl. Med. 2018. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Mayers, J.; Zhen Fan, Z.; Tyacke, R.; Venkataraman, A.; Femminella, G.D.; Perneczky, R.; Gunn, R.N.; Rabiner, E.A.; Matthews, P.M. Evaluation of novel astrocytte marker [11C]BU99008 PET in Alzheimer’s disease: A dementia platform UK experimental medicine study. In Proceedings of the Alzheimer’s Association International Conference, Chicago, IL, USA, 22–26 July 2018. [Google Scholar]
- Roussakis, A.-A.; Mohamed, M.; Myers, J.; Tyacke, R.; Calsolaro, V.; Femminella, G.; Edison, P.; Nutt, D.; Piccini, P. Astrogliosis in Parkinson’s disease dementia: A preliminary report with brain. In Proceedings of the 5th Congress of the European-Academy-of-Neurology (EAN), Oslo, Norway, 29 June–2 July 2019. [Google Scholar]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Tyacke, R.J.; Polychronis, S.; Myers, J.; Gunn, R.N.; Rabiner, E.A.; Nutt, D.; Politis, M. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: An in vivo11C-BU99008 PET study. Brain 2019, 142, 3116–3128. [Google Scholar] [CrossRef]
- Fan, Z.; Calsolaro, V.; Mayers, J.; Tyacke, R.; Venkataraman, A.; Femminella, G.; Perneczky, R.; Gunn, R.; Rabiner, E.; Matthews, P.; et al. Relationship between astrocyte activation using [11C]BU99008 PET, glucose metabolism and amyloid in Alzheimer’s disease: A Dementia Platform UK experimental medicine study. Alzheimer Dement. 2018, 14, 1640. [Google Scholar] [CrossRef]
- Lewis, K.A.; Su, Y.; Jou, O.; Ritchie, C.; Foong, C.; Hynan, L.S.; White, C.L., 3rd; Thomas, P.J.; Hatanpaa, K.J. Abnormal neurites containing C-terminally truncated alpha-synuclein are present in Alzheimer’s disease without conventional Lewy body pathology. Am. J. Pathol 2010, 177, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compta, Y.; Parkkinen, L.; O’Sullivan, S.S.; Vandrovcova, J.; Holton, J.L.; Collins, C.; Lashley, T.; Kallis, C.; Williams, D.R.; de Silva, R.; et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 2011, 134, 1493–1505. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Gomperts, S.N.; Marquie, M.; Locascio, J.J.; Bayer, S.; Johnson, K.A.; Growdon, J.H. PET Radioligands Reveal the Basis of Dementia in Parkinson’s Disease and Dementia with Lewy Bodies. Neurodegener Dis. 2016, 16, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A.; Attems, J. Challenges of multimorbidity of the aging brain: A critical update. J. Neural Transm. 2015, 122, 505–521. [Google Scholar] [CrossRef]
- Akhtar, R.S.; Xie, S.X.; Chen, Y.J.; Rick, J.; Gross, R.G.; Nasrallah, I.M.; Van Deerlin, V.M.; Trojanowski, J.Q.; Chen-Plotkin, A.S.; Hurtig, H.I.; et al. Regional brain amyloid-beta accumulation associates with domain-specific cognitive performance in Parkinson disease without dementia. PLoS ONE 2017, 12, e0177924. [Google Scholar] [CrossRef] [Green Version]
- Ruffmann, C.; Calboli, F.C.; Bravi, I.; Gveric, D.; Curry, L.K.; de Smith, A.; Pavlou, S.; Buxton, J.L.; Blakemore, A.I.; Takousis, P.; et al. Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol. Appl. NeuroBiol. 2016, 42, 436–450. [Google Scholar] [CrossRef]
- Gomperts, S.N.; Rentz, D.M.; Moran, E.; Becker, J.A.; Locascio, J.J.; Klunk, W.E.; Mathis, C.A.; Elmaleh, D.R.; Shoup, T.; Fischman, A.J.; et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008, 71, 903–910. [Google Scholar] [CrossRef]
- Wang, Y.T.; Edison, P. Tau Imaging in Neurodegenerative Diseases Using Positron Emission Tomography. Curr. Neurol. Neurosci. Rep. 2019, 19, 45. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.; Pagano, G.; Politis, M. Dementia spectrum disorders: Lessons learnt from decades with PET research. J. Neural. Transm. 2019, 126, 233–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Dervenoulas, G.; Politis, M.; Alzheimer’s Disease Neuroimaging Initiative. Magnetic resonance imaging in Alzheimer’s disease and mild cognitive impairment. J. Neurol. 2019, 266, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Valkimadi, P.E.; Pagano, G.; Cousins, O.; Dervenoulas, G.; Politis, M.; Alzheimer’s Disease Neuroimaging, I. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 5424–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzinger, T.L.; Blazey, T.; Jack, C.R., Jr.; Koeppe, R.A.; Su, Y.; Xiong, C.; Raichle, M.E.; Snyder, A.Z.; Ances, B.M.; Bateman, R.J.; et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, E4502–E4509. [Google Scholar] [CrossRef] [Green Version]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.Y.; Maruff, P.; Pietrzak, R.H.; Ames, D.; Ellis, K.A.; Harrington, K.; Lautenschlager, N.T.; Szoeke, C.; Martins, R.N.; Masters, C.L.; et al. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer’s disease. Brain 2014, 137, 221–231. [Google Scholar] [CrossRef]
- Teipel, S.; Drzezga, A.; Grothe, M.J.; Barthel, H.; Chetelat, G.; Schuff, N.; Skudlarski, P.; Cavedo, E.; Frisoni, G.B.; Hoffmann, W.; et al. Multimodal imaging in Alzheimer’s disease: Validity and usefulness for early detection. Lancet Neurol. 2015, 14, 1037–1053. [Google Scholar] [CrossRef]
- Engler, H.; Forsberg, A.; Almkvist, O.; Blomquist, G.; Larsson, E.; Savitcheva, I.; Wall, A.; Ringheim, A.; Langstrom, B.; Nordberg, A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 2006, 129, 2856–2866. [Google Scholar] [CrossRef] [Green Version]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef]
- Pike, K.E.; Savage, G.; Villemagne, V.L.; Ng, S.; Moss, S.A.; Maruff, P.; Mathis, C.A.; Klunk, W.E.; Masters, C.L.; Rowe, C.C. Beta-amyloid imaging and memory in non-demented individuals: Evidence for preclinical Alzheimer’s disease. Brain 2007, 130, 2837–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomperts, S.N.; Locascio, J.J.; Marquie, M.; Santarlasci, A.L.; Rentz, D.M.; Maye, J.; Johnson, K.A.; Growdon, J.H. Brain amyloid and cognition in Lewy body diseases. Mov. Disord. 2012, 27, 965–973. [Google Scholar] [CrossRef]
- Melzer, T.R.; Stark, M.R.; Keenan, R.J.; Myall, D.J.; MacAskill, M.R.; Pitcher, T.L.; Livingston, L.; Grenfell, S.; Horne, K.L.; Young, B.N.; et al. Beta Amyloid Deposition Is Not Associated With Cognitive Impairment in Parkinson’s Disease. Front. Neurol. 2019, 10, 391. [Google Scholar] [CrossRef]
- Xia, C.F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, J.H.; Kim, Y.J.; Lee, H.M.; Lyoo, C.H.; Ryu, Y.H.; Lee, M.S. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016, 87, 375–383. [Google Scholar] [CrossRef]
- Pontecorvo, M.J.; Devous, M.D., Sr.; Navitsky, M.; Lu, M.; Salloway, S.; Schaerf, F.W.; Jennings, D.; Arora, A.K.; McGeehan, A.; Lim, N.C.; et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 2017, 140, 748–763. [Google Scholar] [CrossRef]
- Schonhaut, D.R.; McMillan, C.T.; Spina, S.; Dickerson, B.C.; Siderowf, A.; Devous, M.D., Sr.; Tsai, R.; Winer, J.; Russell, D.S.; Litvan, I.; et al. (18) F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: A multicenter study. Ann. Neurol. 2017, 82, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Niccolini, F.; Wilson, H.; Hirschbichler, S.; Yousaf, T.; Pagano, G.; Whittington, A.; Caminiti, S.P.; Erro, R.; Holton, J.L.; Jaunmuktane, Z.; et al. Disease-related patterns of in vivo pathology in Corticobasal syndrome. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2413–2425. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Scholl, M.; Widner, H.; van Westen, D.; Svenningsson, P.; Hagerstrom, D.; Ohlsson, T.; Jogi, J.; Nilsson, C.; Hansson, O. In vivo retention of (18)F-AV-1451 in corticobasal syndrome. Neurology 2017, 89, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Spina, S.; Schonhaut, D.R.; Boeve, B.F.; Seeley, W.W.; Ossenkoppele, R.; O’Neil, J.P.; Lazaris, A.; Rosen, H.J.; Boxer, A.L.; Perry, D.C.; et al. Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology 2017, 88, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Gomperts, S.N.; Locascio, J.J.; Makaretz, S.J.; Schultz, A.; Caso, C.; Vasdev, N.; Sperling, R.; Growdon, J.H.; Dickerson, B.C.; Johnson, K. Tau Positron Emission Tomographic Imaging in the Lewy Body Diseases. JAMA Neurol. 2016, 73, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S.; Pontecorvo, M.J.; Devous, M.D., Sr.; Lu, M.; Arora, A.K.; Truocchio, S.P.; Aldea, P.; Flitter, M.; Locascio, T.; Devine, M.; et al. Positron Emission Tomography Imaging With [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes. JAMA Neurol. 2020, 77, 829–839. [Google Scholar] [CrossRef]
- Vermeiren, C.; Motte, P.; Viot, D.; Mairet-Coello, G.; Courade, J.P.; Citron, M.; Mercier, J.; Hannestad, J.; Gillard, M. The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases. Mov. Disord. 2018, 33, 273–281. [Google Scholar] [CrossRef]
- Ng, K.P.; Pascoal, T.A.; Mathotaarachchi, S.; Therriault, J.; Kang, M.S.; Shin, M.; Guiot, M.C.; Guo, Q.; Harada, R.; Comley, R.A.; et al. Monoamine oxidase B inhibitor, selegiline, reduces (18)F-THK5351 uptake in the human brain. Alzheimers Res. 2017, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Declercq, L.; Rombouts, F.; Koole, M.; Fierens, K.; Marien, J.; Langlois, X.; Andres, J.I.; Schmidt, M.; Macdonald, G.; Moechars, D.; et al. Preclinical Evaluation of (18)F-JNJ64349311, a Novel PET Tracer for Tau Imaging. J. Nucl. Med. 2017, 58, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Bao, W.; Li, M.; Li, L.; Zhang, Z.; Alberts, I.; Brendel, M.; Cumming, P.; Lu, H.; Xiao, Z.; et al. Associations of [(18)F]-APN-1607 Tau PET Binding in the Brain of Alzheimer’s Disease Patients With Cognition and Glucose Metabolism. Front. Neurosci. 2020, 14, 604. [Google Scholar] [CrossRef]
- Hostetler, E.D.; Walji, A.M.; Zeng, Z.; Miller, P.; Bennacef, I.; Salinas, C.; Connolly, B.; Gantert, L.; Haley, H.; Holahan, M.; et al. Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J. Nucl. Med. 2016, 57, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Pascoal, T.A.; Shin, M.; Kang, M.S.; Chamoun, M.; Chartrand, D.; Mathotaarachchi, S.; Bennacef, I.; Therriault, J.; Ng, K.P.; Hopewell, R.; et al. In vivo quantification of neurofibrillary tangles with [(18)F]MK-6240. Alzheimers Res. 2018, 10, 74. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Therriault, J.; Benedet, A.L.; Savard, M.; Lussier, F.Z.; Chamoun, M.; Tissot, C.; Qureshi, M.N.I.; Kang, M.S.; Mathotaarachchi, S.; et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 2020, 143, 2818–2830. [Google Scholar] [CrossRef]
- Sanabria Bohorquez, S.; Marik, J.; Ogasawara, A.; Tinianow, J.N.; Gill, H.S.; Barret, O.; Tamagnan, G.; Alagille, D.; Ayalon, G.; Manser, P.; et al. [(18)F]GTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2077–2089. [Google Scholar] [CrossRef]
- Mueller, A.; Bullich, S.; Barret, O.; Madonia, J.; Berndt, M.; Papin, C.; Perrotin, A.; Koglin, N.; Kroth, H.; Pfeifer, A.; et al. Tau PET imaging with (18)F-PI-2620 in Patients with Alzheimer Disease and Healthy Controls: A First-in-Humans Study. J. Nucl. Med. 2020, 61, 911–919. [Google Scholar] [CrossRef]
- Gobbi, L.C.; Knust, H.; Korner, M.; Honer, M.; Czech, C.; Belli, S.; Muri, D.; Edelmann, M.R.; Hartung, T.; Erbsmehl, I.; et al. Identification of Three Novel Radiotracers for Imaging Aggregated Tau in Alzheimer’s Disease with Positron Emission Tomography. J. Med. Chem. 2017, 60, 7350–7370. [Google Scholar] [CrossRef]
- Honer, M.; Gobbi, L.; Knust, H.; Kuwabara, H.; Muri, D.; Koerner, M.; Valentine, H.; Dannals, R.F.; Wong, D.F.; Borroni, E. Preclinical Evaluation of (18)F-RO6958948, (11)C-RO6931643, and (11)C-RO6924963 as Novel PET Radiotracers for Imaging Tau Aggregates in Alzheimer Disease. J. Nucl. Med. 2018, 59, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Leuzy, A.; Smith, R.; Ossenkoppele, R.; Santillo, A.; Borroni, E.; Klein, G.; Ohlsson, T.; Jogi, J.; Palmqvist, S.; Mattsson-Carlgren, N.; et al. Diagnostic Performance of RO948 F 18 Tau Positron Emission Tomography in the Differentiation of Alzheimer Disease From Other Neurodegenerative Disorders. JAMA Neurol. 2020, 77, 955–965. [Google Scholar] [CrossRef]
- Brendel, M.; Barthel, H.; van Eimeren, T.; Marek, K.; Beyer, L.; Song, M.; Palleis, C.; Gehmeyr, M.; Fietzek, U.; Respondek, G.; et al. Assessment of 18F-PI-2620 as a Biomarker in Progressive Supranuclear Palsy. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Niethammer, M.; Eidelberg, D. Metabolic brain networks in translational neurology: Concepts and applications. Ann. Neurol. 2012, 72, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Schindlbeck, K.A.; Vo, A.; Nguyen, N.; Tang, C.C.; Niethammer, M.; Dhawan, V.; Brandt, V.; Saunders-Pullman, R.; Bressman, S.B.; Eidelberg, D. LRRK2 and GBA Variants Exert Distinct Influences on Parkinson’s Disease-Specific Metabolic Networks. Cereb. Cortex 2020, 30, 2867–2878. [Google Scholar] [CrossRef]
- Franzmeier, N.; Neitzel, J.; Rubinski, A.; Smith, R.; Strandberg, O.; Ossenkoppele, R.; Hansson, O.; Ewers, M.; Alzheimer’s Disease Neuroimaging Initiative. Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat. Commun. 2020, 11, 347. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [Green Version]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef]
- Schapira, A.H. Targeting mitochondria for neuroprotection in Parkinson’s disease. Antioxid Redox Signal. 2012, 16, 965–973. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. NeuroBiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.W.; Bandyopadhyay, U.; Jiang, Y.; et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011, 7, 788–789. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, C.; Crosio, C.; Vitale, C.; Sanna, G.; Carri, M.T.; Barone, P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum. Mol. Genet. 2007, 16, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.L.; Chou, A.H.; Yeh, T.H.; Li, A.H.; Chen, Y.L.; Kuo, Y.L.; Tsai, S.R.; Yu, S.T. PINK1 mutants associated with recessive Parkinson’s disease are defective in inhibiting mitochondrial release of cytochrome c. NeuroBiol. Dis. 2007, 28, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Kawarai, T.; Paitel, E.; Sanjo, N.; Maj, M.; Scheid, M.; Chen, F.; Gu, Y.; Hasegawa, H.; Salehi-Rad, S.; et al. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J. Biol. Chem. 2005, 280, 34025–34032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and inflammatory neurodegeneration. Biochem. Soc. Trans. 2007, 35, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef]
- Day, M.; Wang, Z.; Ding, J.; An, X.; Ingham, C.A.; Shering, A.F.; Wokosin, D.; Ilijic, E.; Sun, Z.; Sampson, A.R.; et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006, 9, 251–259. [Google Scholar] [CrossRef]
- Bellucci, A.; Mercuri, N.B.; Venneri, A.; Faustini, G.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P. Review: Parkinson’s disease: From synaptic loss to connectome dysfunction. Neuropathol. Appl NeuroBiol. 2016, 42, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Plomann, M.; Brundin, P. Huntington’s disease: A synaptopathy? Trends Mol. Med. 2003, 9, 414–420. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [Green Version]
- Kaniyappan, S.; Chandupatla, R.R.; Mandelkow, E.M.; Mandelkow, E. Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimers Dement. 2017, 13, 1270–1291. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Stoothoff, W.; Jones, P.B.; Spires-Jones, T.L.; Joyner, D.; Chhabra, E.; Bercury, K.; Fan, Z.; Xie, H.; Bacskai, B.; Edd, J.; et al. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 2009, 111, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Menkes-Caspi, N.; Yamin, H.G.; Kellner, V.; Spires-Jones, T.L.; Cohen, D.; Stern, E.A. Pathological tau disrupts ongoing network activity. Neuron 2015, 85, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neuroTransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Vogl, C.; Tanifuji, S.; Danis, B.; Daniels, V.; Foerch, P.; Wolff, C.; Whalley, B.J.; Mochida, S.; Stephens, G.J. Synaptic vesicle glycoprotein 2A modulates vesicular release and calcium channel function at peripheral sympathetic synapses. Eur. J. Neurosci. 2015, 41, 398–409. [Google Scholar] [CrossRef]
- Rabiner, E.A. Imaging Synaptic Density: A Different Look at Neurologic Diseases. J. Nucl. Med. 2018, 59, 380–381. [Google Scholar] [CrossRef] [Green Version]
- Matuskey, D.; Tinaz, S.; Wilcox, K.C.; Naganawa, M.; Toyonaga, T.; Dias, M.; Henry, S.; Pittman, B.; Ropchan, J.; Nabulsi, N.; et al. Synaptic Changes in Parkinson Disease Assessed with in vivo Imaging. Ann. Neurol. 2020, 87, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delva, A.; Van Weehaeghe, D.; Koole, M.; Van Laere, K.; Vandenberghe, W. Loss of Presynaptic Terminal Integrity in the Substantia Nigra in Early Parkinson’s Disease. Mov. Disord. 2020, 35. [Google Scholar] [CrossRef]
- Holland, N.; Jones, P.S.; Savulich, G.; Wiggins, J.K.; Hong, Y.T.; Fryer, T.D.; Manavaki, R.; Sephton, S.M.; Boros, I.; Malpetti, M.; et al. Synaptic Loss in Primary Tauopathies Revealed by [(11) C]UCB-J Positron Emission Tomography. Mov. Disord. 2020, 35. [Google Scholar] [CrossRef]
- Chen, M.K.; Mecca, A.P.; Naganawa, M.; Finnema, S.J.; Toyonaga, T.; Lin, S.F.; Najafzadeh, S.; Ropchan, J.; Lu, Y.; McDonald, J.W.; et al. Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 2018, 75, 1215–1224. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Mecca, A.; Chen, M.-K.; Naganawa, M.; Toyonaga, T.; Godek, T.; Harris, J.; Bartlett, H.; Zhao, W.; Gallezot, J.-D.; Nabulsi, N.; et al. Entorhinal cortical tau accumulation is inversely associated with hippocampal synaptic density in older individuals with normal cognition and early Alzheimer’s disease. In Proceedings of the Human Amyloid Imaging, Miami, FL, USA, 15–17 January 2020. [Google Scholar]
- DiFilippo, A.; Murali, D.; McKinney, G.; Davenport, N.; Barnhart, T.; Engle, J.; Betthauser, T.; Johnson, S.; Bendlin, B.; Christian, B. Preliminary evaluation of synaptic vesicle protein SV2A imaging with [11C]UCB-J across the cognitive spectrum. In Proceedings of the Human Amyloid Imaging, Miami, FL, USA, 15–17 January 2020. [Google Scholar]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Chandra, A.; Niccolini, F.; Esposito, M.; Gunn, R.; Ricciardi, L.; Rabiner, E.; Aarsland, D.; et al. The role of Synaptic vesicle protein 2A (SV2A) in patients with Parkinson’s disease dementia and Dementia with Lewy bodies: An in vivo [11C]UCB-J PET study [abstract]. In Proceedings of the 2019 International Congress of Parkinson’s Disease and Movement Disorders, Nice, France, 22–26 September 2019. [Google Scholar]
- Stevenson, J.; Chamoun, M.; Pascoal, T.A.; Benedet, A.; Kang, M.S.; Mathotaarachchi, S.; Therriault, J.; Thomas, E.; Savard, M.; Tissot, C.; et al. Monitoring disease pathophysiology using multiparametric PET acquisitions: The McGill TRIAD Cohort. In Proceedings of the Human Amyloid Imaging, Miami, FL, USA, 15–17 January 2020. [Google Scholar]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Li, X.; Li, Y.; Wang, C.; Wang, T.; Liu, G.; Baskys, A.; Ueda, K.; Chan, P.; Yu, S. alpha-Synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. J. Neurochem. 2011, 119, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, W.; Li, X.; Li, X.; Yang, H.; Xu, Z.; Yu, S. alpha-Synuclein-induced internalization of NMDA receptors in hippocampal neurons is associated with reduced inward current and Ca(2+) influx upon NMDA stimulation. Neuroscience 2015, 300, 297–306. [Google Scholar] [CrossRef]
- Diogenes, M.J.; Dias, R.B.; Rombo, D.M.; Vicente Miranda, H.; Maiolino, F.; Guerreiro, P.; Nasstrom, T.; Franquelim, H.G.; Oliveira, L.M.; Castanho, M.A.; et al. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.; Jeong, H.; Kwon, J.; Heo, H.Y.; Kwon, J.J.; Yun, H.J.; Kim, C.H.; Han, B.S.; Tong, Y.; Shen, J.; et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008, 314, 2055–2065. [Google Scholar] [CrossRef]
- Piccoli, G.; Condliffe, S.B.; Bauer, M.; Giesert, F.; Boldt, K.; De Astis, S.; Meixner, A.; Sarioglu, H.; Vogt-Weisenhorn, D.M.; Wurst, W.; et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 2011, 31, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.J.; Liu, F.; Pristupa, Z.B.; Niznik, H.B. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001, 15, 916–926. [Google Scholar] [CrossRef]
- Sidhu, A.; Wersinger, C.; Vernier, P. alpha-Synuclein regulation of the dopaminergic transporter: A possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 2004, 565, 1–5. [Google Scholar] [CrossRef]
- Wersinger, C.; Jeannotte, A.; Sidhu, A. Attenuation of the norepinephrine transporter activity and trafficking via interactions with alpha-synuclein. Eur. J. Neurosci. 2006, 24, 3141–3152. [Google Scholar] [CrossRef]
- Wersinger, C.; Rusnak, M.; Sidhu, A. Modulation of the trafficking of the human serotonin transporter by human alpha-synuclein. Eur. J. Neurosci. 2006, 24, 55–64. [Google Scholar] [CrossRef]
- Daggett, L.P.; Sacaan, A.I.; Akong, M.; Rao, S.P.; Hess, S.D.; Liaw, C.; Urrutia, A.; Jachec, C.; Ellis, S.B.; Dreessen, J.; et al. Molecular and functional characterization of recombinant human metabotropic glutamate receptor subtype 5. Neuropharmacology 1995, 34, 871–886. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Crimins, J.L.; Rocher, A.B.; Luebke, J.I. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 2012, 124, 777–795. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.G.; Rowe, J.B. Neurotransmitter deficits from frontotemporal lobar degeneration. Brain 2018, 141, 1263–1285. [Google Scholar] [CrossRef]
- Roy, R.; Niccolini, F.; Pagano, G.; Politis, M. Cholinergic imaging in dementia spectrum disorders. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, T.; Fukushi, K.; Akimoto, Y.; Tamagami, H.; Nozaki, T. Design and evaluation of radioactive acetylcholine analogs for mapping brain acetylcholinesterase (AchE) in vivo. Nucl. Med. Biol. 1994, 21, 801–808. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.; Davis, J.G.; Constantine, G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cognitive correlates of alterations in acetylcholinesterase in Alzheimer’s disease. Neurosci. Lett. 2005, 380, 127–132. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.J.; Constantine, G.M.; Mathis Ch, A.; Davis, J.G.; Moore, R.Y.; Dekosky, S.T. Cognitive correlates of cortical cholinergic denervation in Parkinson’s disease and parkinsonian dementia. J. Neurol. 2006, 253, 242–247. [Google Scholar] [CrossRef]
- Shimada, H.; Hirano, S.; Shinotoh, H.; Aotsuka, A.; Sato, K.; Tanaka, N.; Ota, T.; Asahina, M.; Fukushi, K.; Kuwabara, S.; et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 2009, 73, 273–278. [Google Scholar] [CrossRef]
- Klein, J.C.; Eggers, C.; Kalbe, E.; Weisenbach, S.; Hohmann, C.; Vollmar, S.; Baudrexel, S.; Diederich, N.J.; Heiss, W.D.; Hilker, R. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 2010, 74, 885–892. [Google Scholar] [CrossRef]
- Sabri, O.; Kendziorra, K.; Wolf, H.; Gertz, H.J.; Brust, P. Acetylcholine receptors in dementia and mild cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging 2008, 35 (Suppl. 1), S30–S45. [Google Scholar] [CrossRef]
- Meyer, P.M.; Strecker, K.; Kendziorra, K.; Becker, G.; Hesse, S.; Woelpl, D.; Hensel, A.; Patt, M.; Sorger, D.; Wegner, F.; et al. Reduced alpha4beta2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Arch. Gen. Psychiatry 2009, 66, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, D.L.; Markesbery, W.R. Altered serotonergic and cholinergic synaptic markers in Pick’s disease. Arch. Neurol. 1991, 48, 796–799. [Google Scholar] [CrossRef]
- Hirano, S.; Shinotoh, H.; Shimada, H.; Aotsuka, A.; Tanaka, N.; Ota, T.; Sato, K.; Ito, H.; Kuwabara, S.; Fukushi, K.; et al. Cholinergic imaging in corticobasal syndrome, progressive supranuclear palsy and frontotemporal dementia. Brain 2010, 133, 2058–2068. [Google Scholar] [CrossRef] [Green Version]
- Gilman, S.; Koeppe, R.A.; Nan, B.; Wang, C.N.; Wang, X.; Junck, L.; Chervin, R.D.; Consens, F.; Bhaumik, A. Cerebral cortical and subcortical cholinergic deficits in parkinsonian syndromes. Neurology 2010, 74, 1416–1423. [Google Scholar] [CrossRef] [Green Version]
- Candy, J.M.; Perry, R.H.; Perry, E.K.; Irving, D.; Blessed, G.; Fairbairn, A.F.; Tomlinson, B.E. Pathological changes in the nucleus of Meynert in Alzheimer’s and Parkinson’s diseases. J. Neurol. Sci. 1983, 59, 277–289. [Google Scholar] [CrossRef]
- Perry, E.K.; Curtis, M.; Dick, D.J.; Candy, J.M.; Atack, J.R.; Bloxham, C.A.; Blessed, G.; Fairbairn, A.; Tomlinson, B.E.; Perry, R.H. Cholinergic correlates of cognitive impairment in Parkinson’s disease: Comparisons with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1985, 48, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.D.; Brogan, D.; Mirra, S.S. The nucleus basalis of Meynert in neurological disease: A quantitative morphological study. Ann. Neurol. 1985, 17, 163–170. [Google Scholar] [CrossRef]
- Schulz, J.; Pagano, G.; Fernandez Bonfante, J.A.; Wilson, H.; Politis, M. Nucleus basalis of Meynert degeneration precedes and predicts cognitive impairment in Parkinson’s disease. Brain 2018, 141, 1501–1516. [Google Scholar] [CrossRef] [Green Version]
- Petrou, M.; Frey, K.A.; Kilbourn, M.R.; Scott, P.J.; Raffel, D.M.; Bohnen, N.I.; Muller, M.L.; Albin, R.L.; Koeppe, R.A. In vivo imaging of human cholinergic nerve terminals with (-)-5-(18)F-fluoroethoxybenzovesamicol: Biodistribution, dosimetry, and tracer kinetic analyses. J. Nucl. Med. 2014, 55, 396–404. [Google Scholar] [CrossRef] [Green Version]
- Donat, C.K.; Hansen, H.H.; Hansen, H.D.; Mease, R.C.; Horti, A.G.; Pomper, M.G.; L’Estrade, E.T.; Herth, M.M.; Peters, D.; Knudsen, G.M.; et al. In Vitro and In Vivo Characterization of Dibenzothiophene Derivatives [(125)I]Iodo-ASEM and [(18)F]ASEM as Radiotracers of Homo- and Heteromeric alpha7 Nicotinic Acetylcholine Receptors. Molecules 2020, 25, 1425. [Google Scholar] [CrossRef] [Green Version]
- Horti, A.G.; Gao, Y.; Kuwabara, H.; Wang, Y.; Abazyan, S.; Yasuda, R.P.; Tran, T.; Xiao, Y.; Sahibzada, N.; Holt, D.P.; et al. 18F-ASEM, a radiolabeled antagonist for imaging the alpha7-nicotinic acetylcholine receptor with PET. J. Nucl. Med. 2014, 55, 672–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Koros, C.; Yousaf, T.; Picillo, M.; Polychronis, S.; Simitsi, A.; Giordano, B.; Chappell, Z.; et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T alpha-synuclein parkinsonism: A cross-sectional study. Lancet Neurol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wile, D.J.; Agarwal, P.A.; Schulzer, M.; Mak, E.; Dinelle, K.; Shahinfard, E.; Vafai, N.; Hasegawa, K.; Zhang, J.; McKenzie, J.; et al. Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: Cross-sectional studies. Lancet Neurol. 2017, 16, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Wu, K.; Loane, C.; Kiferle, L.; Molloy, S.; Brooks, D.J.; Piccini, P. Staging of serotonergic dysfunction in Parkinson’s disease: An in vivo 11C-DASB PET study. NeuroBiol. Dis. 2010, 40, 216–221. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Turkheimer, F.E.; Molloy, S.; Brooks, D.J.; Piccini, P. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology 2010, 75, 1920–1927. [Google Scholar] [CrossRef]
- Wilson, H.; Giordano, B.; Turkheimer, F.E.; Chaudhuri, K.R.; Politis, M. Serotonergic dysregulation is linked to sleep problems in Parkinson’s disease. Neuroimage Clin. 2018, 18, 630–637. [Google Scholar] [CrossRef]
- Pavese, N.; Metta, V.; Bose, S.K.; Chaudhuri, K.R.; Brooks, D.J. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain 2010, 133, 3434–3443. [Google Scholar] [CrossRef]
- Politis, M.; Loane, C.; Wu, K.; Brooks, D.J.; Piccini, P. Serotonergic mediated body mass index changes in Parkinson’s disease. NeuroBiol. Dis. 2011, 43, 609–615. [Google Scholar] [CrossRef]
- Maillet, A.; Krack, P.; Lhommee, E.; Metereau, E.; Klinger, H.; Favre, E.; Le Bars, D.; Schmitt, E.; Bichon, A.; Pelissier, P.; et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain 2016, 139, 2486–2502. [Google Scholar] [CrossRef] [Green Version]
- Ballanger, B.; Strafella, A.P.; van Eimeren, T.; Zurowski, M.; Rusjan, P.M.; Houle, S.; Fox, S.H. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch. Neurol. 2010, 67, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Loane, C.; Wu, K.; Bain, P.; Brooks, D.J.; Piccini, P.; Politis, M. Serotonergic loss in motor circuitries correlates with severity of action-postural tremor in PD. Neurology 2013, 80, 1850–1855. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Wu, K.; Loane, C.; Brooks, D.J.; Kiferle, L.; Turkheimer, F.E.; Bain, P.; Molloy, S.; Piccini, P. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J. Clin. Investig. 2014, 124, 1340–1349. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Piccini, P. Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci. Transl. Med. 2010, 2, 38ra46. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Noristani, H.N.; Verkhratsky, A. The serotonergic system in ageing and Alzheimer’s disease. Prog NeuroBiol. 2012, 99, 15–41. [Google Scholar] [CrossRef]
- Kepe, V.; Barrio, J.R.; Huang, S.C.; Ercoli, L.; Siddarth, P.; Shoghi-Jadid, K.; Cole, G.M.; Satyamurthy, N.; Cummings, J.L.; Small, G.W.; et al. Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2006, 103, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Sgambato-Faure, V.; Billard, T.; Metereau, E.; Duperrier, S.; Fieux, S.; Costes, N.; Tremblay, L.; Zimmer, L. Characterization and Reliability of [(18)F]2FNQ1P in Cynomolgus Monkeys as a PET Radiotracer for Serotonin 5-HT6 Receptors. Front. Pharm. 2017, 8, 471. [Google Scholar] [CrossRef] [Green Version]
- Emery, S.; Fieux, S.; Vidal, B.; Courault, P.; Bouvard, S.; Tourvieille, C.; Iecker, T.; Billard, T.; Zimmer, L.; Lancelot, S. Preclinical validation of [(18)F]2FNQ1P as a specific PET radiotracer of 5-HT6 receptors in rat, pig, non-human primate and human brain tissue. Nucl. Med. Biol. 2020, 82–83, 57–63. [Google Scholar] [CrossRef]
- Franceschi, M.; Anchisi, D.; Pelati, O.; Zuffi, M.; Matarrese, M.; Moresco, R.M.; Fazio, F.; Perani, D. Glucose metabolism and serotonin receptors in the frontotemporal lobe degeneration. Ann. Neurol. 2005, 57, 216–225. [Google Scholar] [CrossRef]
- Turner, M.R.; Rabiner, E.A.; Al-Chalabi, A.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Andersen, P.M. Cortical 5-HT1A receptor binding in patients with homozygous D90A SOD1 vs sporadic ALS. Neurology 2007, 68, 1233–1235. [Google Scholar] [CrossRef]
- Turner, M.R.; Rabiner, E.A.; Hammers, A.; Al-Chalabi, A.; Grasby, P.M.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N. [11C]-WAY100635 PET demonstrates marked 5-HT1A receptor changes in sporadic ALS. Brain 2005, 128, 896–905. [Google Scholar] [CrossRef]
- da Cunha-Bang, S.; Ettrup, A.; Mc Mahon, B.; Skibsted, A.P.; Schain, M.; Lehel, S.; Dyssegaard, A.; Jorgensen, L.M.; Moller, K.; Gillings, N.; et al. Measuring endogenous changes in serotonergic neurotransmission with [(11)C]Cimbi-36 positron emission tomography in humans. Transl. Psychiatry 2019, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Erritzoe, D.; Ashok, A.H.; Searle, G.E.; Colasanti, A.; Turton, S.; Lewis, Y.; Huiban, M.; Moz, S.; Passchier, J.; Saleem, A.; et al. Serotonin release measured in the human brain: A PET study with [(11)C]CIMBI-36 and d-amphetamine challenge. Neuropsychopharmacology 2020, 45, 804–810. [Google Scholar] [CrossRef]
- Ciranna, L. Serotonin as a modulator of glutamate- and GABA-mediated neurotransmission: Implications in physiological functions and in pathology. Curr. Neuropharmacol. 2006, 4, 101–114. [Google Scholar] [CrossRef] [Green Version]
- de Natale, E.R.; Niccolini, F.; Wilson, H.; Politis, M. Molecular Imaging of the Dopaminergic System in Idiopathic Parkinson’s Disease. Int. Rev. NeuroBiol. 2018, 141, 131–172. [Google Scholar] [CrossRef]
- Martorana, A.; Koch, G. Is dopamine involved in Alzheimer’s disease? Front. Aging Neurosci. 2014, 6, 252. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Wu, X.; Acheampong, K.; Liu, A. Dopamine and Dopamine Receptors in Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Borasio, G.D.; Linke, R.; Schwarz, J.; Schlamp, V.; Abel, A.; Mozley, P.D.; Tatsch, K. Dopaminergic deficit in amyotrophic lateral sclerosis assessed with [I-123] IPT single photon emission computed tomography. J. Neurol. Neurosurg. Psychiatry 1998, 65, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Politis, M. Neuroimaging in Parkinson disease: From research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef]
- Stoessl, A.J.; Lehericy, S.; Strafella, A.P. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet 2014, 384, 532–544. [Google Scholar] [CrossRef] [Green Version]
- McNeill, A.; Wu, R.M.; Tzen, K.Y.; Aguiar, P.C.; Arbelo, J.M.; Barone, P.; Bhatia, K.; Barsottini, O.; Bonifati, V.; Bostantjopoulou, S.; et al. Dopaminergic neuronal imaging in genetic Parkinson’s disease: Insights into pathogenesis. PLoS ONE 2013, 8, e69190. [Google Scholar] [CrossRef] [Green Version]
- Barber, T.R.; Klein, J.C.; Mackay, C.E.; Hu, M.T.M. Neuroimaging in pre-motor Parkinson’s disease. Neuroimage Clin. 2017, 15, 215–227. [Google Scholar] [CrossRef]
- Niccolini, F.; Pagano, G.; Fusar-Poli, P.; Wood, A.; Mrzljak, L.; Sampaio, C.; Politis, M. Striatal molecular alterations in HD gene carriers: A systematic review and meta-analysis of PET studies. J. Neurol. Neurosurg. Psychiatry 2018, 89, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Christopher, L.; Marras, C.; Duff-Canning, S.; Koshimori, Y.; Chen, R.; Boileau, I.; Segura, B.; Monchi, O.; Lang, A.E.; Rusjan, P.; et al. Combined insular and striatal dopamine dysfunction are associated with executive deficits in Parkinson’s disease with mild cognitive impairment. Brain 2014, 137, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Bruck, A.; Portin, R.; Lindell, A.; Laihinen, A.; Bergman, J.; Haaparanta, M.; Solin, O.; Rinne, J.O. Positron emission tomography shows that impaired frontal lobe functioning in Parkinson’s disease is related to dopaminergic hypofunction in the caudate nucleus. Neurosci. Lett. 2001, 311, 81–84. [Google Scholar] [CrossRef]
- Jokinen, P.; Bruck, A.; Aalto, S.; Forsback, S.; Parkkola, R.; Rinne, J.O. Impaired cognitive performance in Parkinson’s disease is related to caudate dopaminergic hypofunction and hippocampal atrophy. Parkinsonism Relat. Disord. 2009, 15, 88–93. [Google Scholar] [CrossRef]
- Caspell-Garcia, C.; Simuni, T.; Tosun-Turgut, D.; Wu, I.W.; Zhang, Y.; Nalls, M.; Singleton, A.; Shaw, L.A.; Kang, J.H.; Trojanowski, J.Q.; et al. Multiple modality biomarker prediction of cognitive impairment in prospectively followed de novo Parkinson disease. PLoS ONE 2017, 12, e0175674. [Google Scholar] [CrossRef]
- Schrag, A.; Siddiqui, U.F.; Anastasiou, Z.; Weintraub, D.; Schott, J.M. Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: A cohort study. Lancet Neurol. 2017, 16, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Alves, G.; Bronnick, K.; Aarsland, D.; Blennow, K.; Zetterberg, H.; Ballard, C.; Kurz, M.W.; Andreasson, U.; Tysnes, O.B.; Larsen, J.P.; et al. CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson’s disease: The Norwegian ParkWest study. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1080–1086. [Google Scholar] [CrossRef]
- Yousaf, T.; Pagano, G.; Niccolini, F.; Politis, M. Predicting cognitive decline with non-clinical markers in Parkinson’s disease (PRECODE-2). J. Neurol. 2019, 266, 1203–1210. [Google Scholar] [CrossRef]
- Jang, H.; Jang, Y.K.; Park, S.; Kim, S.E.; Kim, S.J.; Cho, S.H.; Youn, J.; Seo, S.W.; Kim, H.J.; Na, D.L. Presynaptic dopaminergic function in early-onset Alzheimer’s disease: An FP-CIT image study. Neurobiol. Aging 2020, 86, 75–80. [Google Scholar] [CrossRef]
- Paredes-Rodriguez, E.; Vegas-Suarez, S.; Morera-Herreras, T.; De Deurwaerdere, P.; Miguelez, C. The Noradrenergic System in Parkinson’s Disease. Front. Pharm. 2020, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Delaville, C.; Deurwaerdere, P.D.; Benazzouz, A. Noradrenaline and Parkinson’s disease. Front. Syst. Neurosci. 2011, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Sara, S.J. The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 2009, 10, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Aston-Jones, G.; Waterhouse, B. Locus coeruleus: From global projection system to adaptive regulation of behavior. Brain Res. 2016, 1645, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Leanza, G.; Gulino, R.; Zorec, R. Noradrenergic Hypothesis Linking Neurodegeneration-Based Cognitive Decline and Astroglia. Front. Mol. Neurosci. 2018, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Nahimi, A.; Sommerauer, M.; Kinnerup, M.B.; Ostergaard, K.; Winterdahl, M.; Jacobsen, J.; Schacht, A.; Johnsen, B.; Damholdt, M.F.; Borghammer, P.; et al. Noradrenergic Deficits in Parkinson Disease Imaged with (11)C-MeNER. J. Nucl. Med. 2018, 59, 659–664. [Google Scholar] [CrossRef] [Green Version]
- Sommerauer, M.; Fedorova, T.D.; Hansen, A.K.; Knudsen, K.; Otto, M.; Jeppesen, J.; Frederiksen, Y.; Blicher, J.U.; Geday, J.; Nahimi, A.; et al. Evaluation of the noradrenergic system in Parkinson’s disease: An 11C-MeNER PET and neuromelanin MRI study. Brain 2018, 141, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Brumberg, J.; Tran-Gia, J.; Lapa, C.; Isaias, I.U.; Samnick, S. PET imaging of noradrenaline transporters in Parkinson’s disease: Focus on scan time. Ann. Nucl. Med. 2019, 33, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Remy, P.; Doder, M.; Lees, A.; Turjanski, N.; Brooks, D. Depression in Parkinson’s disease: Loss of dopamine and noradrenaline innervation in the limbic system. Brain 2005, 128, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Benussi, A.; Alberici, A.; Buratti, E.; Ghidoni, R.; Gardoni, F.; Di Luca, M.; Padovani, A.; Borroni, B. Toward a Glutamate Hypothesis of Frontotemporal Dementia. Front. Neurosci. 2019, 13, 304. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G.; Mobius, H.J.; Stoffler, A.; Quack, G. Neuroprotective and symptomatological action of memantine relevant for Alzheimer’s disease--a unified glutamatergic hypothesis on the mechanism of action. Neurotox Res. 2000, 2, 85–97. [Google Scholar] [CrossRef]
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. AJNR Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar]
- Mecca, A.P.; McDonald, J.W.; Michalak, H.R.; Godek, T.A.; Harris, J.E.; Pugh, E.A.; Kemp, E.C.; Chen, M.K.; Salardini, A.; Nabulsi, N.B.; et al. PET imaging of mGluR5 in Alzheimer’s disease. Alzheimers Res. 2020, 12, 15. [Google Scholar] [CrossRef]
- Treyer, V.; Gietl, A.F.; Suliman, H.; Gruber, E.; Meyer, R.; Buchmann, A.; Johayem, A.; Unschuld, P.G.; Nitsch, R.M.; Buck, A.; et al. Reduced uptake of [11C]-ABP688, a PET tracer for metabolic glutamate receptor 5 in hippocampus and amygdala in Alzheimer’s dementia. Brain Behav. 2020, 10, e01632. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, M.; Dirkx, N.; Casteels, C.; Laere, K.V. Excitotoxic neurodegeneration is associated with a focal decrease in metabotropic glutamate receptor type 5 availability: An in vivo PET imaging study. Sci. Rep. 2019, 9, 12916. [Google Scholar] [CrossRef] [Green Version]
- Leuzy, A.; Zimmer, E.R.; Dubois, J.; Pruessner, J.; Cooperman, C.; Soucy, J.P.; Kostikov, A.; Schirmaccher, E.; Desautels, R.; Gauthier, S.; et al. In vivo characterization of metabotropic glutamate receptor type 5 abnormalities in behavioral variant FTD. Brain Struct. Funct. 2016, 221, 1387–1402. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Zanotti-Fregonara, P.; Zoghbi, S.S.; Gladding, R.L.; Woock, A.E.; Innis, R.B.; Pike, V.W. Synthesis and evaluation in monkey of [(18)F]4-fluoro-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzami de ([(18)F]FIMX): A promising radioligand for PET imaging of brain metabotropic glutamate receptor 1 (mGluR1). J. Med. Chem. 2013, 56, 9146–9155. [Google Scholar] [CrossRef] [Green Version]
- Fujinaga, M.; Yamasaki, T.; Yui, J.; Hatori, A.; Xie, L.; Kawamura, K.; Asagawa, C.; Kumata, K.; Yoshida, Y.; Ogawa, M.; et al. Synthesis and evaluation of novel radioligands for positron emission tomography imaging of metabotropic glutamate receptor subtype 1 (mGluR1) in rodent brain. J. Med. Chem. 2012, 55, 2342–2352. [Google Scholar] [CrossRef]
- Dalfo, E.; Albasanz, J.L.; Martin, M.; Ferrer, I. Abnormal metabotropic glutamate receptor expression and signaling in the cerebral cortex in diffuse Lewy body disease is associated with irregular alpha-synuclein/phospholipase C (PLCbeta1) interactions. Brain Pathol. 2004, 14, 388–398. [Google Scholar] [CrossRef]
- Hines, R.M.; Hines, D.J.; Houston, C.M.; Mukherjee, J.; Haydon, P.G.; Tretter, V.; Smart, T.G.; Moss, S.J. Disrupting the clustering of GABAA receptor alpha2 subunits in the frontal cortex leads to reduced gamma-power and cognitive deficits. Proc. Natl. Acad. Sci. USA 2013, 110, 16628–16633. [Google Scholar] [CrossRef] [Green Version]
- Frankle, W.G.; Cho, R.Y.; Narendran, R.; Mason, N.S.; Vora, S.; Litschge, M.; Price, J.C.; Lewis, D.A.; Mathis, C.A. Tiagabine increases [11C]flumazenil binding in cortical brain regions in healthy control subjects. Neuropsychopharmacology 2009, 34, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Pascual, B.; Prieto, E.; Arbizu, J.; Marti-Climent, J.M.; Penuelas, I.; Quincoces, G.; Zarauza, R.; Pappata, S.; Masdeu, J.C. Decreased carbon-11-flumazenil binding in early Alzheimer’s disease. Brain 2012, 135, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Foster, N.L.; Minoshima, S.; Johanns, J.; Little, R.; Heumann, M.L.; Kuhl, D.E.; Gilman, S. PET measures of benzodiazepine receptors in progressive supranuclear palsy. Neurology 2000, 54, 1768–1773. [Google Scholar] [CrossRef]
- Holthoff, V.A.; Koeppe, R.A.; Frey, K.A.; Penney, J.B.; Markel, D.S.; Kuhl, D.E.; Young, A.B. Positron emission tomography measures of benzodiazepine receptors in Huntington’s disease. Ann. Neurol. 1993, 34, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Kunig, G.; Leenders, K.L.; Sanchez-Pernaute, R.; Antonini, A.; Vontobel, P.; Verhagen, A.; Gunther, I. Benzodiazepine receptor binding in Huntington’s disease: [11C]flumazenil uptake measured using positron emission tomography. Ann. Neurol. 2000, 47, 644–648. [Google Scholar] [CrossRef]
- Turner, M.R.; Hammers, A.; Al-Chalabi, A.; Shaw, C.E.; Andersen, P.M.; Brooks, D.J.; Leigh, P.N. Distinct cerebral lesions in sporadic and ‘D90A’ SOD1 ALS: Studies with [11C]flumazenil PET. Brain 2005, 128, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.R.; Osei-Lah, A.D.; Hammers, A.; Al-Chalabi, A.; Shaw, C.E.; Andersen, P.M.; Brooks, D.J.; Leigh, P.N.; Mills, K.R. Abnormal cortical excitability in sporadic but not homozygous D90A SOD1 ALS. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1279–1285. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Kress, B.T.; Weber, H.J.; Thiyagarajan, M.; Wang, B.; Deane, R.; Benveniste, H.; Iliff, J.J.; Nedergaard, M. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J. Transl. Med. 2013, 11, 107. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Nedergaard, M. Is there a cerebral lymphatic system? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Murchison, C.F.; Westaway, S.K.; Simon, M.J.; Erten-Lyons, D.; Kaye, J.A.; Quinn, J.F.; Iliff, J.J. The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimers Dement. (N. Y.) 2017, 3, 348–359. [Google Scholar] [CrossRef]
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Abeta-amyloid burden. Transl. Psychiatry 2018, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Farrell, C.; Wilson, H.; Dervenoulas, G.; Natale, E.R.D.; Politis, M.; Initiative, A.s.D.N. Aquaporin-4 polymorphisms predict amyloid burden and clinical outcome in the Alzheimer’s disease spectrum. Neurobiol. Aging 2020, 97, 1–9. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nakamura, Y.; Yamada, K.; Huber, V.J.; Tsujita, M.; Nakada, T. Aquaporin-4 positron emission tomography imaging of the human brain: First report. J. Neuroimaging Off. J. Am. Soc. Neuroimaging 2013, 23, 219–223. [Google Scholar] [CrossRef]
- Eide, P.K.; Ringstad, G. MRI with intrathecal MRI gadolinium contrast medium administration: A possible method to assess glymphatic function in human brain. Acta Radiol Open 2015, 4, 2058460115609635. [Google Scholar] [CrossRef]
- Ringstad, G.; Vatnehol, S.A.S.; Eide, P.K. Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain 2017, 140, 2691–2705. [Google Scholar] [CrossRef] [Green Version]
- Eide, P.K.; Ringstad, G. Delayed clearance of cerebrospinal fluid tracer from entorhinal cortex in idiopathic normal pressure hydrocephalus: A glymphatic magnetic resonance imaging study. J. Cereb. Blood Flow Metab. 2019, 39, 1355–1368. [Google Scholar] [CrossRef]
- Ringstad, G.; Valnes, L.M.; Dale, A.M.; Pripp, A.H.; Vatnehol, S.S.; Emblem, K.E.; Mardal, K.A.; Eide, P.K. Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Jaraj, D.; Rabiei, K.; Marlow, T.; Jensen, C.; Skoog, I.; Wikkelso, C. Prevalence of idiopathic normal-pressure hydrocephalus. Neurology 2014, 82, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, V.; Koivisto, A.M.; Savolainen, S.; Rummukainen, J.; Tamminen, J.N.; Tillgren, T.; Vainikka, S.; Pyykko, O.T.; Molsa, J.; Fraunberg, M.; et al. Amyloid and tau proteins in cortical brain biopsy and Alzheimer’s disease. Ann. Neurol. 2010, 68, 446–453. [Google Scholar] [CrossRef]
- Yatsushiro, S.; Sunohara, S.; Hayashi, N.; Hirayama, A.; Matsumae, M.; Atsumi, H.; Kuroda, K. Cardiac-driven Pulsatile Motion of Intracranial Cerebrospinal Fluid Visualized Based on a Correlation Mapping Technique. Magn. Reson. Med. Sci. 2018, 17, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Isoda, H.; Hirano, M.; Takeda, H.; Inagawa, S.; Takehara, Y.; Alley, M.T.; Markl, M.; Pelc, N.J.; Sakahara, H. Visualization of hemodynamics in intracranial arteries using time-resolved three-dimensional phase-contrast MRI. J. Magn. Reson. Imaging 2007, 25, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Hasiloglu, Z.I.; Albayram, S.; Gorucu, Y.; Selcuk, H.; Cagil, E.; Erdemli, H.E.; Adaletli, I. Assessment of CSF flow dynamics using PC-MRI in spontaneous intracranial hypotension. Headache 2012, 52, 808–819. [Google Scholar] [CrossRef]
- Battal, B.; Kocaoglu, M.; Bulakbasi, N.; Husmen, G.; Tuba Sanal, H.; Tayfun, C. Cerebrospinal fluid flow imaging by using phase-contrast MR technique. Br. J. Radiol. 2011, 84, 758–765. [Google Scholar] [CrossRef] [Green Version]
- Odeen, H.; Uppman, M.; Markl, M.; Spottiswoode, B.S. Assessing cerebrospinal fluid flow connectivity using 3D gradient echo phase contrast velocity encoded MRI. Physiol. Meas. 2011, 32, 407–421. [Google Scholar] [CrossRef]
- Hayashi, N.; Matsumae, M.; Yatsushiro, S.; Hirayama, A.; Abdullah, A.; Kuroda, K. Quantitative Analysis of Cerebrospinal Fluid Pressure Gradients in Healthy Volunteers and Patients with Normal Pressure Hydrocephalus. Neurol. Med. Chir. (Tokyo) 2015, 55, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Matsumae, M.; Hirayama, A.; Atsumi, H.; Yatsushiro, S.; Kuroda, K. Velocity and pressure gradients of cerebrospinal fluid assessed with magnetic resonance imaging. J. Neurosurg. 2014, 120, 218–227. [Google Scholar] [CrossRef]
- Rivera-Rivera, L.A.; Turski, P.; Johnson, K.M.; Hoffman, C.; Berman, S.E.; Kilgas, P.; Rowley, H.A.; Carlsson, C.M.; Johnson, S.C.; Wieben, O. 4D flow MRI for intracranial hemodynamics assessment in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 1718–1730. [Google Scholar] [CrossRef]
- Kyrtsos, C.R.; Baras, J.S. Modeling the Role of the Glymphatic Pathway and Cerebral Blood Vessel Properties in Alzheimer’s Disease Pathogenesis. PLoS ONE 2015, 10, e0139574. [Google Scholar] [CrossRef] [Green Version]
- Kiviniemi, V.; Wang, X.; Korhonen, V.; Keinanen, T.; Tuovinen, T.; Autio, J.; LeVan, P.; Keilholz, S.; Zang, Y.F.; Hennig, J.; et al. Ultra-fast magnetic resonance encephalography of physiological brain activity—Glymphatic pulsation mechanisms? J. Cereb. Blood Flow Metab. 2016, 36, 1033–1045. [Google Scholar] [CrossRef]
- Myllyla, T.; Harju, M.; Korhonen, V.; Bykov, A.; Kiviniemi, V.; Meglinski, I. Assessment of the dynamics of human glymphatic system by near-infrared spectroscopy. J. Biophotonics 2018, 11, e201700123. [Google Scholar] [CrossRef]
- Ohene, Y.; Harrison, I.F.; Nahavandi, P.; Ismail, O.; Bird, E.V.; Ottersen, O.P.; Nagelhus, E.A.; Thomas, D.L.; Lythgoe, M.F.; Wells, J.A. Non-invasive MRI of brain clearance pathways using multiple echo time arterial spin labelling: An aquaporin-4 study. Neuroimage 2019, 188, 515–523. [Google Scholar] [CrossRef]
- Taoka, T.; Masutani, Y.; Kawai, H.; Nakane, T.; Matsuoka, K.; Yasuno, F.; Kishimoto, T.; Naganawa, S. Evaluation of glymphatic system activity with the diffusion MR technique: Diffusion tensor image analysis along the perivascular space (DTI-ALPS) in Alzheimer’s disease cases. Jpn. J. Radiol. 2017, 35, 172–178. [Google Scholar] [CrossRef]
- Harrison, I.F.; Siow, B.; Akilo, A.B.; Evans, P.G.; Ismail, O.; Ohene, Y.; Nahavandi, P.; Thomas, D.L.; Lythgoe, M.F.; Wells, J.A. Non-invasive imaging of CSF-mediated brain clearance pathways via assessment of perivascular fluid movement with diffusion tensor MRI. Elife 2018, 7. [Google Scholar] [CrossRef]
- Hou, Y.; Park, S.H.; Wang, Q.; Zhang, J.; Zong, X.; Lin, W.; Shen, D. Enhancement of Perivascular Spaces in 7 T MR Image using Haar Transform of Non-local Cubes and Block-matching Filtering. Sci. Rep. 2017, 7, 8569. [Google Scholar] [CrossRef]
- Potter, G.M.; Chappell, F.M.; Morris, Z.; Wardlaw, J.M. Cerebral perivascular spaces visible on magnetic resonance imaging: Development of a qualitative rating scale and its observer reliability. Cereb. Dis. 2015, 39, 224–231. [Google Scholar] [CrossRef]
- Banerjee, G.; Kim, H.J.; Fox, Z.; Jager, H.R.; Wilson, D.; Charidimou, A.; Na, H.K.; Na, D.L.; Seo, S.W.; Werring, D.J. MRI-visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain 2017, 140, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Shams, S.; Martola, J.; Charidimou, A.; Larvie, M.; Granberg, T.; Shams, M.; Kristoffersen-Wiberg, M.; Wahlund, L.O. Topography and Determinants of Magnetic Resonance Imaging (MRI)-Visible Perivascular Spaces in a Large Memory Clinic Cohort. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Hansen, T.P.; Cain, J.; Thomas, O.; Jackson, A. Dilated perivascular spaces in the Basal Ganglia are a biomarker of small-vessel disease in a very elderly population with dementia. Ajnr Am. J. Neuroradiol. 2015, 36, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.; Berezuk, C.; McNeely, A.A.; Scott, C.J.; Gao, F.; Black, S.E. Visible Virchow-Robin spaces on magnetic resonance imaging of Alzheimer’s disease patients and normal elderly from the Sunnybrook Dementia Study. J. Alzheimers Dis. 2015, 43, 415–424. [Google Scholar] [CrossRef]
- Ratner, V.; Gao, Y.; Lee, H.; Elkin, R.; Nedergaard, M.; Benveniste, H.; Tannenbaum, A. Cerebrospinal and interstitial fluid transport via the glymphatic pathway modeled by optimal mass transport. Neuroimage 2017, 152, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Ratner, V.; Zhu, L.; Kolesov, I.; Nedergaard, M.; Benveniste, H.; Tannenbaum, A. Optimal-mass-transfer-based estimation of glymphatic transport in living brain. Proc. Spie Int. Soc. Opt. Eng. 2015, 9413. [Google Scholar] [CrossRef] [Green Version]
- Linninger, A.A.; Tsakiris, C.; Zhu, D.C.; Xenos, M.; Roycewicz, P.; Danziger, Z.; Penn, R. Pulsatile cerebrospinal fluid dynamics in the human brain. IEEE Trans. Biomed. Eng. 2005, 52, 557–565. [Google Scholar] [CrossRef]
- Linninger, A.A.; Xenos, M.; Sweetman, B.; Ponkshe, S.; Guo, X.; Penn, R. A mathematical model of blood, cerebrospinal fluid and brain dynamics. J. Math. Biol. 2009, 59, 729–759. [Google Scholar] [CrossRef]
- Zhu, D.C.; Xenos, M.; Linninger, A.A.; Penn, R.D. Dynamics of lateral ventricle and cerebrospinal fluid in normal and hydrocephalic brains. J. Magn. Reson. Imaging 2006, 24, 756–770. [Google Scholar] [CrossRef]
- Metzger, F.; Mischek, D.; Stoffers, F. The Connected Steady State Model and the Interdependence of the CSF Proteome and CSF Flow Characteristics. Front. Neurosci. 2017, 11, 241. [Google Scholar] [CrossRef]
- Achariyar, T.M.; Li, B.; Peng, W.; Verghese, P.B.; Shi, Y.; McConnell, E.; Benraiss, A.; Kasper, T.; Song, W.; Takano, T.; et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener 2016, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, K.E.; Koscik, R.L.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Okonkwo, O.C.; Sager, M.A.; Asthana, S.; Johnson, S.C.; Benca, R.M.; et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 2017, 89, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.E.; McLeland, J.S.; Toedebusch, C.D.; Xiong, C.; Fagan, A.M.; Duntley, S.P.; Morris, J.C.; Holtzman, D.M. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013, 70, 587–593. [Google Scholar] [CrossRef]
- Berezuk, C.; Ramirez, J.; Gao, F.; Scott, C.J.; Huroy, M.; Swartz, R.H.; Murray, B.J.; Black, S.E.; Boulos, M.I. Virchow-Robin Spaces: Correlations with Polysomnography-Derived Sleep Parameters. Sleep 2015, 38, 853–858. [Google Scholar] [CrossRef]
- Shokri-Kojori, E.; Wang, G.J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl Acad Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Sing, H.; Belenky, G.; Holcomb, H.; Mayberg, H.; Dannals, R.; Wagner, H.; Thorne, D.; Popp, K.; Rowland, L.; et al. Neural basis of alertness and cognitive performance impairments during sleepiness. I. Effects of 24 h of sleep deprivation on waking human regional brain activity. J. Sleep Res. 2000, 9, 335–352. [Google Scholar] [CrossRef] [Green Version]
- Sprecher, K.E.; Bendlin, B.B.; Racine, A.M.; Okonkwo, O.C.; Christian, B.T.; Koscik, R.L.; Sager, M.A.; Asthana, S.; Johnson, S.C.; Benca, R.M. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. NeuroBiol. Aging 2015, 36, 2568–2576. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.M.; Rainey-Smith, S.R.; Villemagne, V.L.; Weinborn, M.; Bucks, R.S.; Sohrabi, H.R.; Laws, S.M.; Taddei, K.; Macaulay, S.L.; Ames, D.; et al. The Relationship between Sleep Quality and Brain Amyloid Burden. Sleep 2016, 39, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.E.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef]
- Volkow, N.D.; Tomasi, D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Logan, J.; Benveniste, H.; Kim, R.; Thanos, P.K.; Ferre, S. Evidence that sleep deprivation downregulates dopamine D2R in ventral striatum in the human brain. J. Neurosci. 2012, 32, 6711–6717. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Tomasi, D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Wang, R.L.; Logan, J.; Wong, C.; Jayne, M.; Swanson, J.M. Hyperstimulation of striatal D2 receptors with sleep deprivation: Implications for cognitive impairment. Neuroimage 2009, 45, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Logan, J.; Wong, C.; Ma, J.; Pradhan, K.; Tomasi, D.; Thanos, P.K.; et al. Sleep deprivation decreases binding of [11C]raclopride to dopamine D2/D3 receptors in the human brain. J. Neurosci. 2008, 28, 8454–8461. [Google Scholar] [CrossRef]
- Elmenhorst, D.; Kroll, T.; Matusch, A.; Bauer, A. Sleep deprivation increases cerebral serotonin 2A receptor binding in humans. Sleep 2012, 35, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Huang, L.L.; Chen, J.H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal. Transduct Target. 2019, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Chiba-Falek, O.; Lutz, M.W. Towards precision medicine in Alzheimer’s disease: Deciphering genetic data to establish informative biomarkers. Expert Rev. Precis. Med. Drug. Dev. 2017, 2, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Leonenko, G.; Sims, R.; Shoai, M.; Frizzati, A.; Bossu, P.; Spalletta, G.; Fox, N.C.; Williams, J.; The GERAD Consortium; Hardy, J.; et al. Polygenic risk and hazard scores for Alzheimer’s disease prediction. Ann. Clin. Transl. Neurol. 2019, 6, 456–465. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Licher, S.; Leening, M.J.G.; Yilmaz, P.; Wolters, F.J.; Heeringa, J.; Bindels, P.J.E.; Alzheimer’s Disease Neuroimaging Initiative; Vernooij, M.W.; Stephan, B.C.M.; Steyerberg, E.W.; et al. Development and Validation of a Dementia Risk Prediction Model in the General Population: An Analysis of Three Longitudinal Studies. Am. J. Psychiatry 2019, 176, 543–551. [Google Scholar] [CrossRef]
- Walters, K.; Hardoon, S.; Petersen, I.; Iliffe, S.; Omar, R.Z.; Nazareth, I.; Rait, G. Predicting dementia risk in primary care: Development and validation of the Dementia Risk Score using routinely collected data. BMC Med. 2016, 14, 6. [Google Scholar] [CrossRef] [Green Version]
- Solomon, A.; Soininen, H. Dementia: Risk prediction models in dementia prevention. Nat. Rev. Neurol. 2015, 11, 375–377. [Google Scholar] [CrossRef]
- Kivipelto, M.; Ngandu, T.; Laatikainen, T.; Winblad, B.; Soininen, H.; Tuomilehto, J. Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet Neurol. 2006, 5, 735–741. [Google Scholar] [CrossRef]
- Vuoksimaa, E.; Rinne, J.O.; Lindgren, N.; Heikkila, K.; Koskenvuo, M.; Kaprio, J. Middle age self-report risk score predicts cognitive functioning and dementia in 20-40 years. Alzheimers Dement. (Amst.) 2016, 4, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ogrodnik, M.; Devine, S.; Auerbach, S.; Wolf, P.A.; Au, R. Practical risk score for 5-, 10-, and 20-year prediction of dementia in elderly persons: Framingham Heart Study. Alzheimers Dement. 2018, 14, 35–42. [Google Scholar] [CrossRef]
- Barnes, D.E.; Beiser, A.S.; Lee, A.; Langa, K.M.; Koyama, A.; Preis, S.R.; Neuhaus, J.; McCammon, R.J.; Yaffe, K.; Seshadri, S.; et al. Development and validation of a brief dementia screening indicator for primary care. Alzheimers Dement. 2014, 10, 656–665.e1. [Google Scholar] [CrossRef] [Green Version]
- Niethammer, M.; Tang, C.C.; Vo, A.; Nguyen, N.; Spetsieris, P.; Dhawan, V.; Ma, Y.; Small, M.; Feigin, A.; During, M.J.; et al. Gene therapy reduces Parkinson’s disease symptoms by reorganizing functional brain connectivity. Sci. Transl Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Cencioni, C.; Spallotta, F.; Martelli, F.; Valente, S.; Mai, A.; Zeiher, A.M.; Gaetano, C. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 2013, 14, 17643–17663. [Google Scholar] [CrossRef] [Green Version]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef] [Green Version]
- Whittington, A.; Gunn, R.N.; Alzheimer’s Disease Neuroimaging Initiative. Amyloid Load: A More Sensitive Biomarker for Amyloid Imaging. J. Nucl. Med. 2019, 60, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Whittington, A.; Seibyl, J.; Hesterman, J.; Gunn, R. Tau(IQ)—A quantitative algorithm for tau PET imaging in clinical trials. In Proceedings of the 29th International Symposium on Cerebral Blood Flow, Metabolism and Function, Yokohama, Japan, 4–7 July 2019. [Google Scholar]
Figure 1.
Schematic illustration of interlinked genotypes, molecular pathways and clinical phenotypes across age-related neurodegenerative diseases showing the overlap between various components and pathways at different levels, from genetics and molecular pathways to clinical phenotypes. Disentangling this etiological puzzle of known and yet unknown pathways acting distinctly or in concert could improve the stratification of patients into clinical trials and, potentially, help to drive the treatment landscape towards the precision medicine paradigm. The relationship between clinical diagnosis and clinical phenotypes was adapted from Ahmed et al., 2016 [3]. Abbreviations: AD: Alzheimer’s disease, ALS: Amyotrophic Lateral Sclerosis, FTD: Fronto-Temporal Dementia and PD: Parkinson’s disease.
Figure 1.
Schematic illustration of interlinked genotypes, molecular pathways and clinical phenotypes across age-related neurodegenerative diseases showing the overlap between various components and pathways at different levels, from genetics and molecular pathways to clinical phenotypes. Disentangling this etiological puzzle of known and yet unknown pathways acting distinctly or in concert could improve the stratification of patients into clinical trials and, potentially, help to drive the treatment landscape towards the precision medicine paradigm. The relationship between clinical diagnosis and clinical phenotypes was adapted from Ahmed et al., 2016 [3]. Abbreviations: AD: Alzheimer’s disease, ALS: Amyotrophic Lateral Sclerosis, FTD: Fronto-Temporal Dementia and PD: Parkinson’s disease.

Figure 2.
Overview of molecular pathways targeted with PET radioligands. (A) Mitochondrial dysfunction and energy dysregulation can be investigated using (18F)BCPP-EF, for mitochondrial complex 1 and (11C)SA4503 for sigma 1 receptor. (B) Neuroinflammation can be investigated by targeting translator protein expressed on activated microglia using PET radioligands such as (11C)PK11195 and astroglia activation using novel PET radioligands such as (11C)BU99008 for imidazoline 2-binding sites. (C) Abnormal protein aggregation of tau and amyloid-β can be quantified using specific radioligands such as (18F)AV1451 and (18F)Florbetaben, respectively. (D) Synaptic pathology can be investigated using (11C)UCB-J targeting synaptic vesicle glycoprotein 2A. (E) Dysregulation of neurotransmitter systems can be investigated by employing various PET radioligands, including serotonergic markers such as (11C)DASB for the serotonin transporter and dopaminergic markers such as presynaptic markers (18F)DOPA for dopamine storage, (11C)PE2I for dopamine transporter and (11C)Raclopride for postsynaptic dopaminergic receptors, as well as PET radioligands for noradrenergic, glutamatergic and GABAergic systems. Abbreviations: D2R/D3R: Dopamine type-2/type-3 receptor, DAT: Dopamine transporter, DDC: Dopa Decarboxylase, I2BS: Imidazoline 2-binding sites, MC1: Mitochondrial Complex 1, SERT: Serotonin transporter, Sigma 1R: Sigma 1 receptor, SV2A: Synaptic vesicle glycoprotein 2A and TSPO: Translocator protein.
Figure 2.
Overview of molecular pathways targeted with PET radioligands. (A) Mitochondrial dysfunction and energy dysregulation can be investigated using (18F)BCPP-EF, for mitochondrial complex 1 and (11C)SA4503 for sigma 1 receptor. (B) Neuroinflammation can be investigated by targeting translator protein expressed on activated microglia using PET radioligands such as (11C)PK11195 and astroglia activation using novel PET radioligands such as (11C)BU99008 for imidazoline 2-binding sites. (C) Abnormal protein aggregation of tau and amyloid-β can be quantified using specific radioligands such as (18F)AV1451 and (18F)Florbetaben, respectively. (D) Synaptic pathology can be investigated using (11C)UCB-J targeting synaptic vesicle glycoprotein 2A. (E) Dysregulation of neurotransmitter systems can be investigated by employing various PET radioligands, including serotonergic markers such as (11C)DASB for the serotonin transporter and dopaminergic markers such as presynaptic markers (18F)DOPA for dopamine storage, (11C)PE2I for dopamine transporter and (11C)Raclopride for postsynaptic dopaminergic receptors, as well as PET radioligands for noradrenergic, glutamatergic and GABAergic systems. Abbreviations: D2R/D3R: Dopamine type-2/type-3 receptor, DAT: Dopamine transporter, DDC: Dopa Decarboxylase, I2BS: Imidazoline 2-binding sites, MC1: Mitochondrial Complex 1, SERT: Serotonin transporter, Sigma 1R: Sigma 1 receptor, SV2A: Synaptic vesicle glycoprotein 2A and TSPO: Translocator protein.


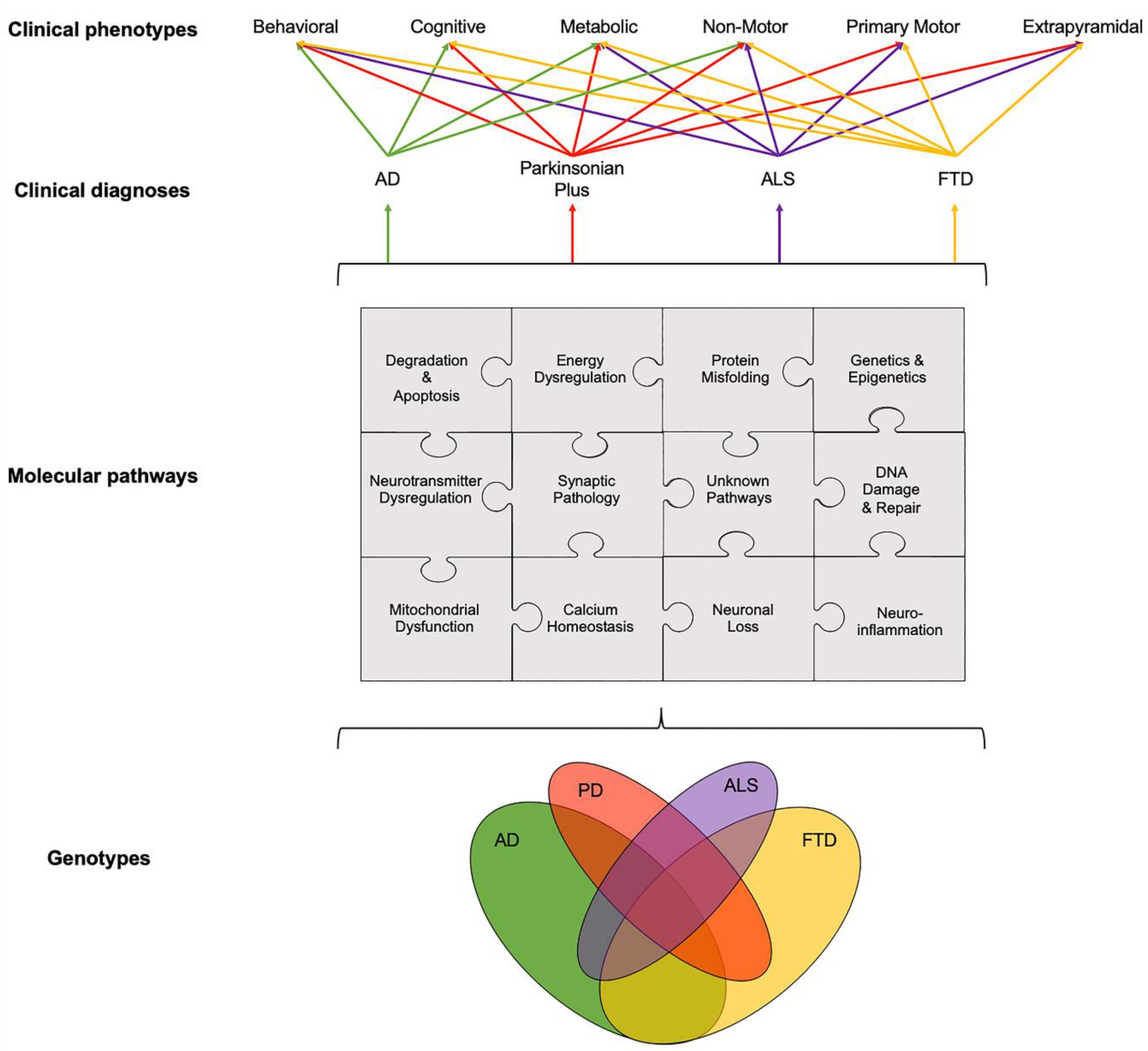{kind=link}
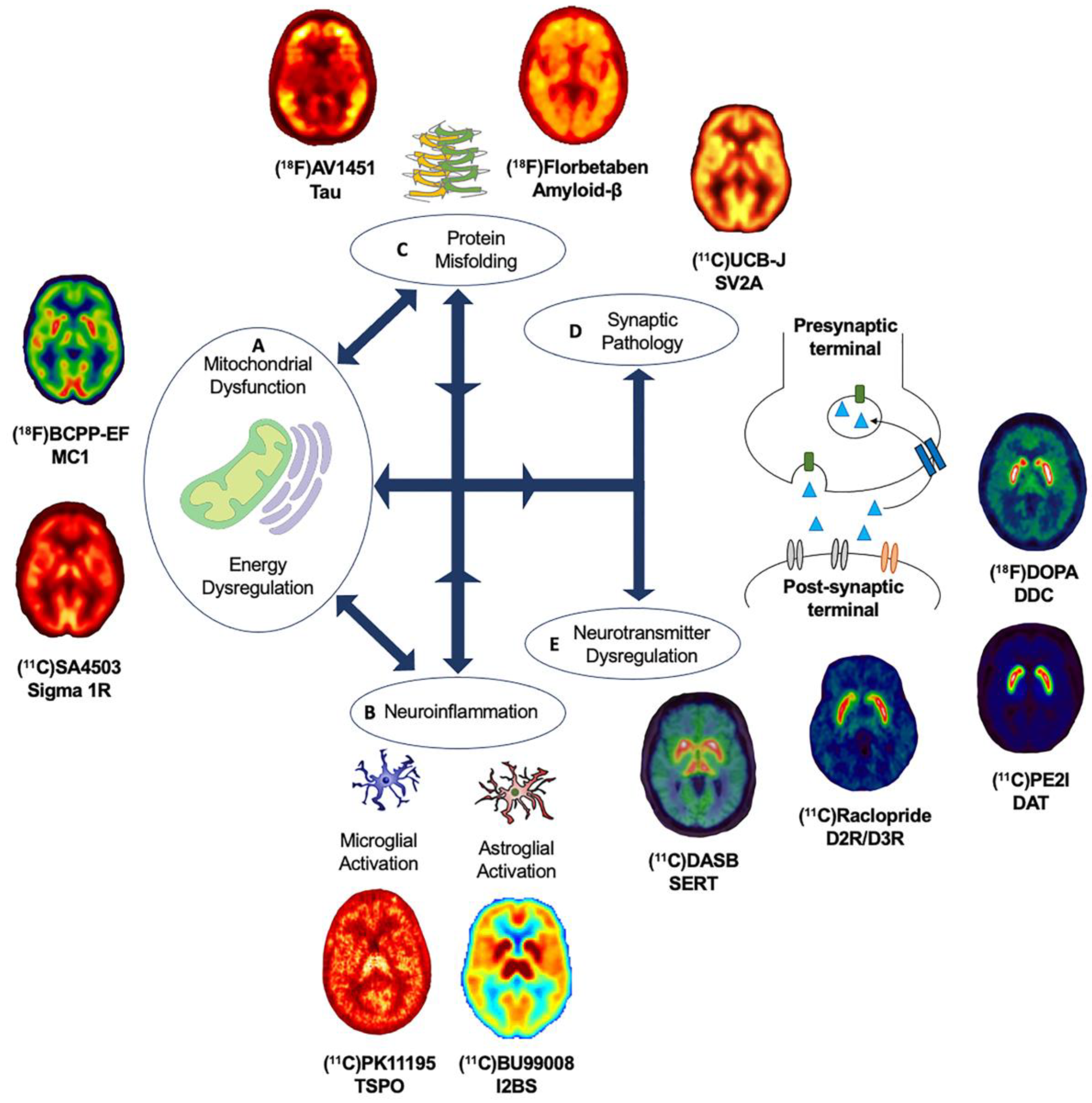{kind=link}
Table 1.
Overview of new PET radioligand for novel targets and related molecular pathways across age-related neurodegenerative diseases.
Table 1.
Overview of new PET radioligand for novel targets and related molecular pathways across age-related neurodegenerative diseases.
| PET Radioligand | Target | Molecular Pathway |
|---|---|---|
| Protein aggregation | ||
| (18F)PI-2620 (18F)MK-2640 (18F)RO-948 (18F)GTPI (18F)JNJ64349311 (18F)APN-1607 | Tau | Tau deposition |
| Neurotransmitter Dysregulation | ||
| (11C)Cimbi-36 | 5-HT2R | Serotonergic system |
| (18F)2FNQ1P | 5-HT6R | |
| (18F)FEOBV (18F)VAT | VAChT | Cholinergic system |
| (18F)ASEM | α7-nAChR | |
| (11C)ABP688 (11C)AZD9272 (11C)FPEB | mGluR5 | Glutamatergic system |
| (11C)ITMM (18F)FIMX | mGluR1 | |
| (18F)ΜΝΙ-444 | A2A | Adrenergic system |
| (11C)MK-8278 (11C)GSK189254 | H3R | Histaminergic system |
| Neuroinflammation | ||
| (18F)DPA-714 (11C)ER176 | TSPO | Microglial activation |
| (11C)BU99008 | I2BS | Astroglial activation |
| (11C)JNJ717 (11C)SMW139 (18F)JNJ-64413739 (11C)GSK1482160 | P2X7 | Purinoceptors |
| (11C)PS13 | COX-1 | Cyclooxygenase |
| (11C)MC1 | COX-2 | |
| Synaptic Pathology | ||
| (11C)UCB-J (18F)UCB-H | SV2A | Synaptic function |
| Mitochondrial dysfunction and energy dysregulation | ||
| (18F)BCPP-EF | MC1 | Mitochondrial function |
| (11C)SA-4503 | σ-1R | Mitochondrial-associated membrane |
| Epigenetics | ||
| (11C)Martinostat | HDAC | Epigenetics |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wilson, H.; Politis, M.; Rabiner, E.A.; Middleton, L.T. Novel PET Biomarkers to Disentangle Molecular Pathways across Age-Related Neurodegenerative Diseases. Cells 2020, 9, 2581. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122581
AMA Style
Wilson H, Politis M, Rabiner EA, Middleton LT. Novel PET Biomarkers to Disentangle Molecular Pathways across Age-Related Neurodegenerative Diseases. Cells. 2020; 9(12):2581. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122581
Chicago/Turabian StyleWilson, Heather, Marios Politis, Eugenii A. Rabiner, and Lefkos T. Middleton. 2020. "Novel PET Biomarkers to Disentangle Molecular Pathways across Age-Related Neurodegenerative Diseases" Cells 9, no. 12: 2581. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122581
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.