The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives



Abstract
:1. Introduction
2. AML Heterogeneity and Prognostic Evaluation
2.1. Prognostic Evaluation as the Basis for Therapeutic Strategies
2.1.1. Non-Relapse Mortality Due to Intensive Antileukemic Therapy
2.1.2. The Factors Currently Used for Evaluation of Relapse Risk in Patients with Newly Diagnosed AML
2.1.3. Prognostic Evaluation of Patients with Relapsed AML
2.1.4. Prognostic Evaluation of Patients Receiving AML Stabilizing Treatment
2.2. Improved Prognostic Evaluation and More Extensive Molecular Characterization of Therapeutic Targets Are Needed in AML
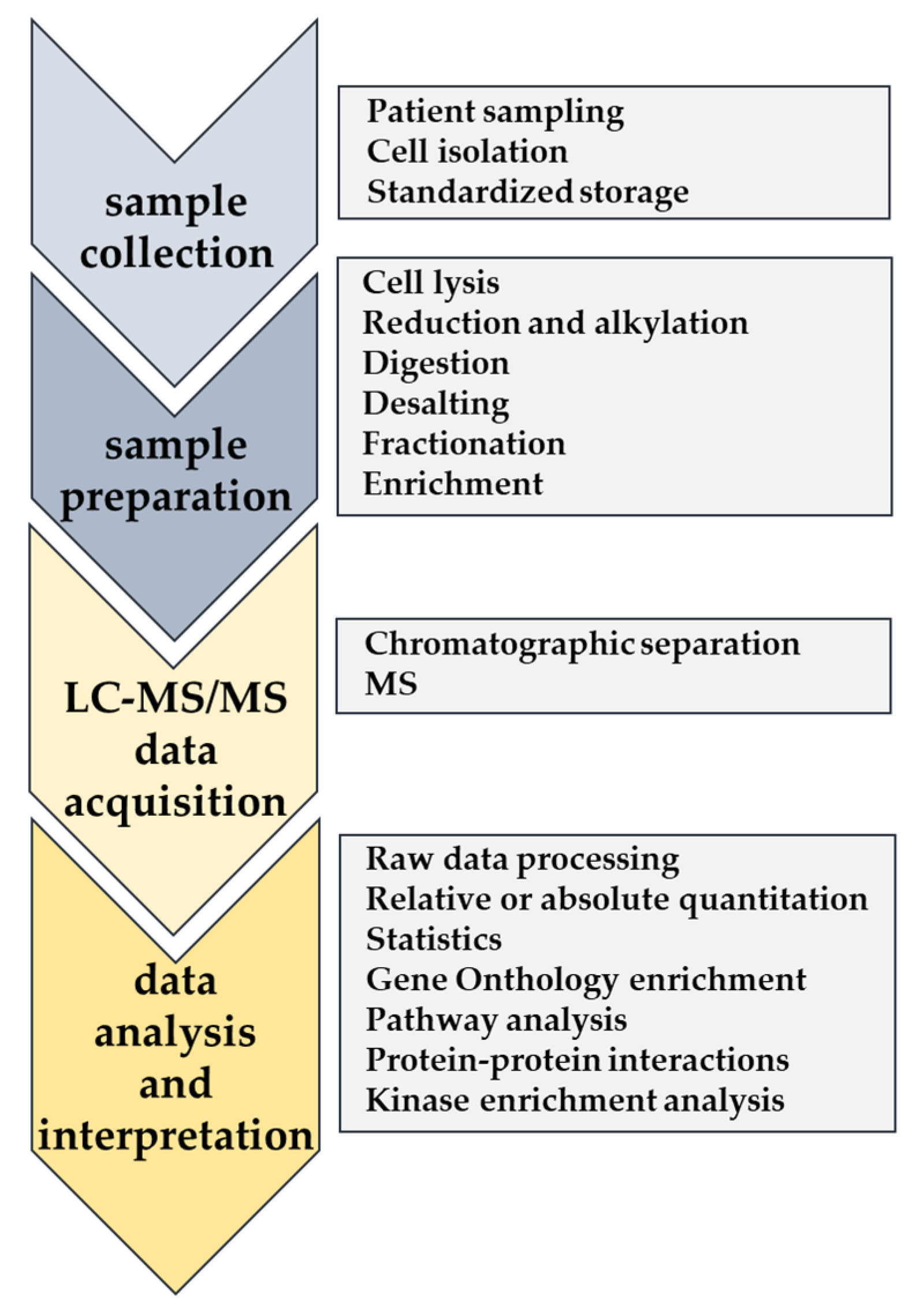
3. Recent Advances in LC-MS/MS-Based Proteomics and Phosphoproteomics for AML Blast Cells
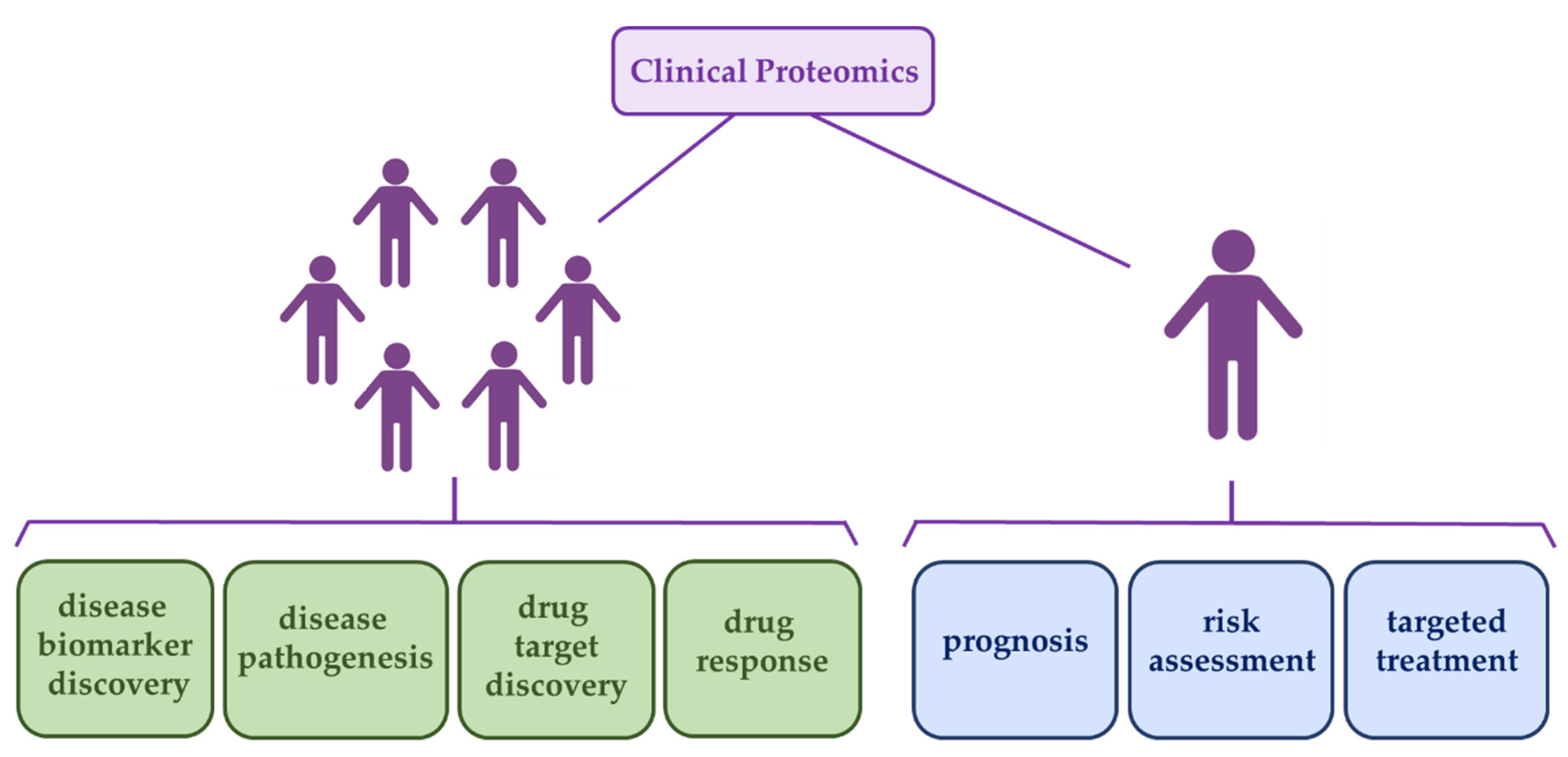
4. Implementation of Proteomic Workflows in Cancer Clinical Routines
4.1. Advanced Proteomic Workflows with Potential Clinical Use
4.2. Bioinformatics Pipelines with Potential Clinical Use
5. Current Status on the Implementation of Proteomic Workflows in AML Clinical Routines
6. Other Considerations in Clinical AML Proteomics
7. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cohen Freue, G.V.; Meredith, A.; Smith, D.; Bergman, A.; Sasaki, M.; Lam, K.K.; Hollander, Z.; Opushneva, N.; Takhar, M.; Lin, D.; et al. Computational biomarker pipeline from discovery to clinical implementation: Plasma proteomic biomarkers for cardiac transplantation. PLoS Comput. Biol. 2013, 9, e1002963. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.; Broom, B.M.; Verhaak, R.G.W.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S.; Goldberg, R.M.; Lenz, H.J.; Shields, A.F.; Gibney, G.T.; Tan, A.R.; Brown, J.; Eisenberg, B.; Heath, E.I.; Phuphanich, S.; et al. The current state of molecular testing in the treatment of patients with solid tumors, 2019. Cancer J. Clin. 2019, 69, 305–343. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, A.P.; Imyanitov, E.N. Molecular Diagnostics in Clinical Oncology. Front. Mol. Biosci. 2018, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Applying a Proteomics-Based Approach to the Clinic. Available online: https://www.news-medical.net/news/20191120/Applying-a-Proteomics-based-Approach-to-the-Clinic.aspx (accessed on 28 July 2020).
- Huang, T.; Deng, C.X. Current Progresses of Exosomes as Cancer Diagnostic and Prognostic Biomarkers. Int. J. Biol. Sci. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Mardamshina, M.; Geiger, T. Next-Generation Proteomics and Its Application to Clinical Breast Cancer Research. Am. J. Pathol. 2017, 187, 2175–2184. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Mannan, A.; Muhsen, I.N.; Barragan, E.; Sanz, M.A.; Mohty, M.; Hashmi, S.K.; Aljurf, M. Genotypic and Phenotypic Characteristics of Acute Promyelocytic Leukemia Translocation Variants. Hematol. Oncol. Stem. Cell 2020. [Google Scholar] [CrossRef]
- Thomas, X. Acute Promyelocytic Leukemia: A History over 60 Years-From the Most Malignant to the most Curable Form of Acute Leukemia. Oncology 2019, 7, 33–65. [Google Scholar] [CrossRef] [Green Version]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Gratwohl, A.; Schlenk, R.F.; Sierra, J.; Bornhauser, M.; Juliusson, G.; Racil, Z.; Rowe, J.M.; Russell, N.; Mohty, M.; et al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: An integrated-risk adapted approach. Nat. Rev. Clin. Oncol. 2012, 9, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.H. Acute myeloid leukemia: 2014 update on risk-stratification and management. Am. J. Hematol. 2014, 89, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.J.; Janssen, J.J.; van de Loosdrecht, A.A. Risk factors for relapse after allogeneic transplantation in acute myeloid leukemia. Haematologica 2016, 101, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Fredly, H.; Gjertsen, B.T.; Bruserud, O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenetics 2013, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- Wen, B.; You, W.; Yang, S.; Du, X. Indirect comparison of azacitidine and decitabine for the therapy of elderly patients with acute myeloid leukemia: A systematic review and network meta-analysis. Exp. Hematol. Oncol. 2020, 9, 3. [Google Scholar] [CrossRef]
- Reikvam, H.; Hauge, M.; Brenner, A.K.; Hatfield, K.J.; Bruserud, O. Emerging therapeutic targets for the treatment of human acute myeloid leukemia (part 1)—Gene transcription, cell cycle regulation, metabolism and intercellular communication. Expert Rev. Hematol. 2015, 8, 299–313. [Google Scholar] [CrossRef]
- Reikvam, H.; Hoang, T.T.; Bruserud, O. Emerging therapeutic targets in human acute myeloid leukemia (part 2)—Bromodomain inhibition should be considered as a possible strategy for various patient subsets. Expert Rev. Hematol. 2015, 8, 315–327. [Google Scholar] [CrossRef]
- Rowe, J.M. Will new agents impact survival in AML? Best Pr. Res. Clin. Haematol. 2019, 32, 101094. [Google Scholar] [CrossRef] [PubMed]
- Bohl, S.R.; Bullinger, L.; Rucker, F.G. New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. Int. J. Mol. Sci. 2019, 20, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Karp, J.E.; Emadi, A.; Othus, M.; Gale, R.P. Recent drug approvals for newly diagnosed acute myeloid leukemia: Gifts or a Trojan horse? Leukemia 2020, 34, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.; Pautas, C.; Terre, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Mayer, L.D.; Tardi, P.; Louie, A.C. CPX-351: A nanoscale liposomal co-formulation of daunorubicin and cytarabine with unique biodistribution and tumor cell uptake properties. Int. J. Nanomed. 2019, 14, 3819–3830. [Google Scholar] [CrossRef] [Green Version]
- Megias-Vericat, J.E.; Ballesta-Lopez, O.; Barragan, E.; Montesinos, P. IDH1-mutated relapsed or refractory AML: Current challenges and future prospects. Blood Lymphat. Cancer 2019, 9, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. The role of small molecule Flt3 receptor protein-tyrosine kinase inhibitors in the treatment of Flt3-positive acute myelogenous leukemias. Pharm. Res. 2020, 155, 104725. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Talati, C.; Sweet, K. Recently approved therapies in acute myeloid leukemia: A complex treatment landscape. Leuk. Res. 2018, 73, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Davis, R.B.; Schiffer, C.A.; Berg, D.T.; Powell, B.L.; Schulman, P.; Omura, G.A.; Moore, J.O.; McIntyre, O.R.; Frei, E. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N. Engl. J. Med. 1994, 331, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Elsawy, M.; Storer, B.E.; Milano, F.; Sandmaier, B.M.; Delaney, C.; Salit, R.B.; Rashad, A.H.; Woolfrey, A.E.; Appelbaum, F.R.; Storb, R.; et al. Prognostic Performance of the Augmented Hematopoietic Cell Transplantation-Specific Comorbidity/Age Index in Recipients of Allogeneic Hematopoietic Stem Cell Transplantation from Alternative Graft Sources. Biol. Blood Marrow Transplant. 2019, 25, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Borthakur, G.; Ravandi, F.; Faderl, S.; Verstovsek, S.; Thomas, D.; Wierda, W.; Ferrajoli, A.; Kornblau, S.; Pierce, S.; et al. The haematopoietic cell transplantation comorbidity index score is predictive of early death and survival in patients over 60 years of age receiving induction therapy for acute myeloid leukaemia. Br. J. Haematol. 2007, 136, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Salit, R.B.; Oliver, D.C.; Delaney, C.; Sorror, M.L.; Milano, F. Prognostic Value of the Hematopoietic Cell Transplantation Comorbidity Index for Patients Undergoing Reduced-Intensity Conditioning Cord Blood Transplantation. Biol Blood Marrow Transplant. 2017, 23, 654–658. [Google Scholar] [CrossRef] [Green Version]
- Sorror, M.L.; Giralt, S.; Sandmaier, B.M.; De Lima, M.; Shahjahan, M.; Maloney, D.G.; Deeg, H.J.; Appelbaum, F.R.; Storer, B.; Storb, R. Hematopoietic cell transplantation specific comorbidity index as an outcome predictor for patients with acute myeloid leukemia in first remission: Combined FHCRC and MDACC experiences. Blood 2007, 110, 4606–4613. [Google Scholar] [CrossRef] [Green Version]
- Sorror, M.L.; Storb, R.F.; Sandmaier, B.M.; Maziarz, R.T.; Pulsipher, M.A.; Maris, M.B.; Bhatia, S.; Ostronoff, F.; Deeg, H.J.; Syrjala, K.L.; et al. Comorbidity-age index: A clinical measure of biologic age before allogeneic hematopoietic cell transplantation. J. Clin. Oncol. 2014, 32, 3249–3256. [Google Scholar] [CrossRef]
- Sorror, M.L.; Storer, B.E.; Fathi, A.T.; Gerds, A.T.; Medeiros, B.C.; Shami, P.; Brunner, A.M.; Sekeres, M.A.; Mukherjee, S.; Pena, E.; et al. Development and Validation of a Novel Acute Myeloid Leukemia-Composite Model to Estimate Risks of Mortality. JAMA Oncol. 2017, 3, 1675–1682. [Google Scholar] [CrossRef]
- Wais, V.; Bunjes, D.; Kuchenbauer, F.; Sorror, M.L. Comorbidities, age, and other patient-related predictors of allogeneic hematopoietic cell transplantation outcomes. Expert Rev. Hematol. 2018, 11, 805–816. [Google Scholar] [CrossRef]
- Versluis, J.; Labopin, M.; Niederwieser, D.; Socie, G.; Schlenk, R.F.; Milpied, N.; Nagler, A.; Blaise, D.; Rocha, V.; Cornelissen, J.J.; et al. Prediction of non-relapse mortality in recipients of reduced intensity conditioning allogeneic stem cell transplantation with AML in first complete remission. Leukemia 2015, 29, 51–57. [Google Scholar] [CrossRef]
- Gratwohl, A.; Stern, M.; Brand, R.; Apperley, J.; Baldomero, H.; de Witte, T.; Dini, G.; Rocha, V.; Passweg, J.; Sureda, A.; et al. Risk score for outcome after allogeneic hematopoietic stem cell transplantation: A retrospective analysis. Cancer 2009, 115, 4715–4726. [Google Scholar] [CrossRef]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet. 2019, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Handschuh, L. Not Only Mutations Matter: Molecular Picture of Acute Myeloid Leukemia Emerging from Transcriptome Studies. J. Oncol. 2019, 2019, 7239206. [Google Scholar] [CrossRef] [Green Version]
- Moarii, M.; Papaemmanuil, E. Classification and risk assessment in AML: Integrating cytogenetics and molecular profiling. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasebo, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Doskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers Basel. 2020, 12, 709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasebo, E.; Berven, F.S.; Hovland, R.; Doskeland, S.O.; Bruserud, O.; Selheim, F.; Hernandez-Valladares, M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers Basel. 2020, 12, 1466. [Google Scholar] [CrossRef] [PubMed]
- Gronningsaeter, I.S.; Reikvam, H.; Aasebo, E.; Bartaula-Brevik, S.; Tvedt, T.H.; Bruserud, O.; Hatfield, K.J. Targeting Cellular Metabolism in Acute Myeloid Leukemia and The Role of Patient Heterogeneity. Cells 2020, 9, 1155. [Google Scholar] [CrossRef]
- Wheatley, K.; Burnett, A.K.; Goldstone, A.H.; Gray, R.G.; Hann, I.M.; Harrison, C.J.; Rees, J.K.; Stevens, R.F.; Walker, H. A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. United Kingdom Medical Research Council’s Adult and Childhood Leukaemia Working Parties. Br. J. Haematol. 1999, 107, 69–79. [Google Scholar] [CrossRef]
- Hulegardh, E.; Nilsson, C.; Lazarevic, V.; Garelius, H.; Antunovic, P.; Rangert Derolf, A.; Mollgard, L.; Uggla, B.; Wennstrom, L.; Wahlin, A.; et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: A report from the Swedish Acute Leukemia Registry. Am. J. Hematol. 2015, 90, 208–214. [Google Scholar] [CrossRef]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Baron, F.; Stevens-Kroef, M.; Kicinski, M.; Meloni, G.; Muus, P.; Marie, J.P.; Halkes, C.J.M.; Thomas, X.; Vrhovac, R.; Specchia, G.; et al. Cytogenetic clonal heterogeneity is not an independent prognosis factor in 15-60-year-old AML patients: Results on 1291 patients included in the EORTC/GIMEMA AML-10 and AML-12 trials. Ann. Hematol. 2018, 97, 1785–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, B.C.; Othus, M.; Fang, M.; Appelbaum, F.R.; Erba, H.P. Cytogenetic heterogeneity negatively impacts outcomes in patients with acute myeloid leukemia. Haematologica 2015, 100, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Tvedt, T.H.A.; Reikvam, H.; Bruserud, O. Clonal Heterogeneity Reflected by PI3K-AKT-mTOR Signaling in Human Acute Myeloid Leukemia Cells and Its Association with Adverse Prognosis. Cancers 2018, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estey, E. Management of persistent AML at day 14. Best Pr. Res. Clin. Haematol. 2014, 27, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.A.; Wood, B.L.; Othus, M.; Hourigan, C.S.; Ustun, C.; Linden, M.A.; DeFor, T.E.; Malagola, M.; Anthias, C.; Valkova, V.; et al. Minimal residual disease prior to allogeneic hematopoietic cell transplantation in acute myeloid leukemia: A meta-analysis. Haematologica 2017, 102, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [Green Version]
- Bose, P.; Vachhani, P.; Cortes, J.E. Treatment of Relapsed/Refractory Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2017, 18, 17. [Google Scholar] [CrossRef]
- Ramos, N.R.; Mo, C.C.; Karp, J.E.; Hourigan, C.S. Current Approaches in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J. Clin. Med. 2015, 4, 665–695. [Google Scholar] [CrossRef] [Green Version]
- Craddock, C.; Hoelzer, D.; Komanduri, K.V. Current status and future clinical directions in the prevention and treatment of relapse following hematopoietic transplantation for acute myeloid and lymphoblastic leukemia. Bone Marrow Transplant. 2019, 54, 6–16. [Google Scholar] [CrossRef]
- Rautenberg, C.; Germing, U.; Haas, R.; Kobbe, G.; Schroeder, T. Relapse of Acute Myeloid Leukemia after Allogeneic Stem Cell Transplantation: Prevention, Detection, and Treatment. Int. J. Mol. Sci. 2019, 20, 228. [Google Scholar] [CrossRef] [Green Version]
- De Jonge, H.J.; Valk, P.J.; de Bont, E.S.; Schuringa, J.J.; Ossenkoppele, G.; Vellenga, E.; Huls, G. Prognostic impact of white blood cell count in intermediate risk acute myeloid leukemia: Relevance of mutated NPM1 and FLT3-ITD. Haematologica 2011, 96, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhou, L.; Zhang, X.; Tang, B.; Zhu, X.; Liu, H.; Sun, Z.; Zheng, C. Impact Of ELN Risk Stratification, Induction Chemotherapy Regimens And Hematopoietic Stem Cell Transplantation On Outcomes In Hyperleukocytic Acute Myeloid Leukemia With Initial White Blood Cell Count More Than 100 x 10(9)/L. Cancer Manag. Res. 2019, 11, 9495–9503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- How, J.; Sykes, J.; Gupta, V.; Yee, K.W.; Schimmer, A.D.; Schuh, A.C.; Minden, M.D.; Kamel-Reid, S.; Brandwein, J.M. Influence of FLT3-internal tandem duplication allele burden and white blood cell count on the outcome in patients with intermediate-risk karyotype acute myeloid leukemia. Cancer 2012, 118, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Aasebo, E.; Hernandez-Valladares, M.; Tsykunova, G.; Reikvam, H. Therapeutic targeting of leukemic stem cells in acute myeloid leukemia—The biological background for possible strategies. Expert Opin. Drug Discov. 2017, 12, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Aarstad, H.H.; Tvedt, T.H.A. Combined C-Reactive Protein and Novel Inflammatory Parameters as a Predictor in Cancer-What Can We Learn from the Hematological Experience? Cancers Basel. 2020, 12, 1966. [Google Scholar] [CrossRef] [PubMed]
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. [Google Scholar] [CrossRef] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Gronningsaeter, I.S.; Aasebo, E.; Hernandez-Valladares, M.; Hagen, K.M.; Rye, K.P.; Berven, F.S.; Selheim, F.; Reikvam, H.; et al. Effects of insulin and pathway inhibitors on the PI3K-Akt-mTOR phosphorylation profile in acute myeloid leukemia cells. Signal Transduct. Target. 2019, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Zjablovskaja, P.; Florian, M.C. Acute Myeloid Leukemia: Aging and Epigenetics. Cancers 2019, 12, 103. [Google Scholar] [CrossRef] [Green Version]
- Ryningen, A.; Stapnes, C.; Lassalle, P.; Corbascio, M.; Gjertsen, B.T.; Bruserud, O. A subset of patients with high-risk acute myelogenous leukemia shows improved peripheral blood cell counts when treated with the combination of valproic acid, theophylline and all-trans retinoic acid. Leuk. Res. 2009, 33, 779–787. [Google Scholar] [CrossRef]
- Gronningsaeter, I.S.; Fredly, H.K.; Gjertsen, B.T.; Hatfield, K.J.; Bruserud, O. Systemic Metabolomic Profiling of Acute Myeloid Leukemia Patients before and During Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid, Valproic Acid, and Low-Dose Chemotherapy. Cells 2019, 8, 1229. [Google Scholar] [CrossRef] [Green Version]
- Aasebo, E.; Forthun, R.B.; Berven, F.; Selheim, F.; Hernandez-Valladares, M. Global Cell Proteome Profiling, Phospho-signaling and Quantitative Proteomics for Identification of New Biomarkers in Acute Myeloid Leukemia Patients. Curr. Pharm. Biotechnol. 2016, 17, 52–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estey, E.H. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am. J. Hematol. 2013, 88, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.E.; Vener, T.I.; Raponi, M.; Ritchie, E.K.; Smith, B.D.; Gore, S.D.; Morris, L.E.; Feldman, E.J.; Greer, J.M.; Malek, S.; et al. Multi-institutional phase 2 clinical and pharmacogenomic trial of tipifarnib plus etoposide for elderly adults with newly diagnosed acute myelogenous leukemia. Blood 2012, 119, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, O.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In vitro culture of aml cells—The present use in experimental studies and the possible importance for future therapeutic approaches. Stem. Cells 2001, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Vaudel, M.; Selheim, F.; Berven, F.; Bruserud, O. Proteogenomics approaches for studying cancer biology and their potential in the identification of acute myeloid leukemia biomarkers. Expert Rev. Proteom. 2017, 14, 649–663. [Google Scholar] [CrossRef]
- Roboz, J.; Roboz, G.J. Mass spectrometry in leukemia research and treatment. Expert Rev. Hematol. 2015, 8, 225–235. [Google Scholar] [CrossRef]
- Selheim, F.; Aasebo, E.; Ribas, C.; Aragay, A.M. An Overview on G Protein-coupled Receptor-induced Signal Transduction in Acute Myeloid Leukemia. Curr. Med. Chem. 2019, 26, 5293–5316. [Google Scholar] [CrossRef]
- Tong, J.; Helmy, M.; Cavalli, F.M.; Jin, L.; St-Germain, J.; Karisch, R.; Taylor, P.; Minden, M.D.; Taylor, M.D.; Neel, B.G.; et al. Integrated analysis of proteome, phosphotyrosine-proteome, tyrosine-kinome, and tyrosine-phosphatome in acute myeloid leukemia. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- De Boer, B.; Prick, J.; Pruis, M.G.; Keane, P.; Imperato, M.R.; Jaques, J.; Brouwers-Vos, A.Z.; Hogeling, S.M.; Woolthuis, C.M.; Nijk, M.T.; et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 2018, 34, 674–689.e8. [Google Scholar] [CrossRef] [Green Version]
- Aasebo, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.S.; Brenner, A.K.; Bruserud, O. Proteomic Profiling of Primary Human Acute Myeloid Leukemia Cells Does Not Reflect Their Constitutive Release of Soluble Mediators. Proteomes 2018, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forthun, R.B.; Aasebo, E.; Rasinger, J.D.; Bedringaas, S.L.; Berven, F.; Selheim, F.; Bruserud, O.; Gjertsen, B.T. Phosphoprotein DIGE profiles reflect blast differentiation, cytogenetic risk stratification, FLT3/NPM1 mutations and therapy response in acute myeloid leukaemia. J. Proteom. 2018, 173, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Aasebo, E.; Brenner, A.K.; Bartaula-Brevik, S.; Gronningsaeter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, O. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Aasebo, E.; Hernandez-Valladares, M.; Brenner, A.K.; Bartaula-Brevik, S.; Berven, F.; Selheim, F.; Skavland, J.; Gjertsen, B.T.; et al. Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin. Targets. 2018, 22, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.K.; Aasebo, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.; Gronningsaeter, I.S.; Bartaula-Brevik, S.; Bruserud, O. The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome. Cancers 2019, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Raffel, S.; Klimmeck, D.; Falcone, M.; Demir, A.; Pouya, A.; Zeisberger, P.; Lutz, C.; Tinelli, M.; Bischel, O.; Bullinger, L.; et al. Quantitative proteomics reveals specific metabolic features of Acute Myeloid Leukemia stem cells. Blood 2020. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Schmidt, J.R.; Rucker-Braun, E.; Heidrich, K.; von Bonin, M.; Stolzel, F.; Thiede, C.; Middeke, J.M.; Ehninger, G.; Bornhauser, M.; Schetelig, J.; et al. Pilot Study on Mass Spectrometry-Based Analysis of the Proteome of CD34(+)CD123(+) Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia. Proteomes 2018, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Alanazi, B.; Munje, C.R.; Rastogi, N.; Williamson, A.J.K.; Taylor, S.; Hole, P.S.; Hodges, M.; Doyle, M.; Baker, S.; Gilkes, A.F.; et al. Integrated nuclear proteomics and transcriptomics identifies S100A4 as a therapeutic target in acute myeloid leukemia. Leukemia 2020, 34, 427–440. [Google Scholar] [CrossRef]
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 17, 17. [Google Scholar] [CrossRef]
- Manza, L.L.; Stamer, S.L.; Ham, A.J.; Codreanu, S.G.; Liebler, D.C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Ostasiewicz, P.; Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 2011, 10, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.W.; Chang, C.L.; Chen, S.F. Evaluation of peptide fractionation strategies used in proteome analysis. J. Sep. Sci. 2012, 35, 3293–3301. [Google Scholar] [CrossRef]
- Batth, T.S.; Francavilla, C.; Olsen, J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Geiger, T.; Wisniewski, J.R.; Cox, J.; Zanivan, S.; Kruger, M.; Ishihama, Y.; Mann, M. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat. Protoc. 2011, 6, 147–157. [Google Scholar] [CrossRef]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- America, A.H.; Cordewener, J.H. Comparative LC-MS: A landscape of peaks and valleys. Proteomics 2008, 8, 731–749. [Google Scholar] [CrossRef]
- Aasebo, E.; Vaudel, M.; Mjaavatten, O.; Gausdal, G.; Van der Burgh, A.; Gjertsen, B.T.; Doskeland, S.O.; Bruserud, O.; Berven, F.S.; Selheim, F. Performance of super-SILAC based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics 2014, 14, 1971–1976. [Google Scholar] [CrossRef]
- Aasebo, E.; Mjaavatten, O.; Vaudel, M.; Farag, Y.; Selheim, F.; Berven, F.; Bruserud, O.; Hernandez-Valladares, M. Freezing effects on the acute myeloid leukemia cell proteome and phosphoproteome revealed using optimal quantitative workflows. J. Proteom. 2016, 145, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Aasebo, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, O.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thingholm, T.E.; Larsen, M.R. Phosphopeptide Enrichment by Immobilized Metal Affinity Chromatography. Methods Mol. Biol. 2016, 1355, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, T.E.; Jorgensen, T.J.; Jensen, O.N.; Larsen, M.R. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc. 2006, 1, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, T.E.; Jensen, O.N.; Robinson, P.J.; Larsen, M.R. SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol. Cell. Proteom. 2008, 7, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Schaab, C.; Oppermann, F.S.; Klammer, M.; Pfeifer, H.; Tebbe, A.; Oellerich, T.; Krauter, J.; Levis, M.; Perl, A.E.; Daub, H.; et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia 2014, 28, 716–719. [Google Scholar] [CrossRef] [Green Version]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Lowenberg, B.; Zweegman, S.; et al. Leukemic stem cell frequency: A strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef] [Green Version]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Kelder, A.; Cloos, J.; Schuurhuis, G.J. Immunophenotypic Detection of Measurable Residual (Stem Cell) Disease Using LAIP Approach in Acute Myeloid Leukemia. Curr. Protoc. Cytom. 2019, 91, e66. [Google Scholar] [CrossRef] [Green Version]
- Amon, S.; Meier-Abt, F.; Gillet, L.C.; Dimitrieva, S.; Theocharides, A.P.A.; Manz, M.G.; Aebersold, R. Sensitive Quantitative Proteomics of Human Hematopoietic Stem and Progenitor Cells by Data-independent Acquisition Mass Spectrometry. Mol. Cell. Proteom. 2019, 18, 1454–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, V. A dream of single-cell proteomics. Nat. Methods 2019, 16, 809–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coscia, F.; Doll, S.; Bech, J.M.; Schweizer, L.; Mund, A.; Lengyel, E.; Lindebjerg, J.; Madsen, G.I.; Moreira, J.M.; Mann, M. A streamlined mass spectrometry-based proteomics workflow for large-scale FFPE tissue analysis. J. Pathol. 2020, 251, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell. Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsboll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Edfors, F.; Bostrom, T.; Forsstrom, B.; Zeiler, M.; Johansson, H.; Lundberg, E.; Hober, S.; Lehtio, J.; Mann, M.; Uhlen, M. Immunoproteomics Using Polyclonal Antibodies and Stable Isotope-labeled Affinity-purified Recombinant Proteins. Mol. Cell. Proteom. 2014, 13, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Coscia, F.; Lengyel, E.; Duraiswamy, J.; Ashcroft, B.; Bassani-Sternberg, M.; Wierer, M.; Johnson, A.; Wroblewski, K.; Montag, A.; Yamada, S.D.; et al. Multi-level Proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy Target in Ovarian Cancer. Cell 2018, 175, 159–170.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, H.; Penning, R.; Fitzpatrick, M.A.; Garrigues, L.B.; Wu, W.; MacGillavry, H.D.; Hoogenraad, C.C.; Heck, A.J.; Altelaar, A.F. Robust, Sensitive, and Automated Phosphopeptide Enrichment Optimized for Low Sample Amounts Applied to Primary Hippocampal Neurons. J. Proteome Res. 2017, 16, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, P.A.; Markey, S.P.; Roth, J.; Mirokhin, Y.; Yan, X.J.; Tchekhovskoi, D.V.; Edwards, N.J.; Thangudu, R.R.; Ketchum, K.A.; Kinsinger, C.R.; et al. A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Common Data Analysis Pipeline. J. Proteome Res. 2016, 15, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turewicz, M.; Kohl, M.; Ahrens, M.; Mayer, G.; Uszkoreit, J.; Naboulsi, W.; Bracht, T.; Megger, D.A.; Sitek, B.; Marcus, K.; et al. BioInfra.Prot: A comprehensive proteomics workflow including data standardization, protein inference, expression analysis and data publication. J. Biotechnol. 2017, 261, 116–125. [Google Scholar] [CrossRef]
- Eisenacher, M.; (Ruhr Universitat Bochum, Germany). Personal Communication, 2020.
- Rost, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Slagel, J.; Sun, Z.; Moritz, R.L. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteom. Clin. Appl. 2015, 9, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Cox, J.; (Max Planck Institute of Biochemistry, Martinsried, Germany). Personal Communication, 2020.
- Guldbrandsen, A.; Farag, Y.; Kroksveen, A.C.; Oveland, E.; Lereim, R.R.; Opsahl, J.A.; Myhr, K.M.; Berven, F.S.; Barsnes, H. CSF-PR 2.0: An Interactive Literature Guide to Quantitative Cerebrospinal Fluid Mass Spectrometry Data from Neurodegenerative Disorders. Mol. Cell. Proteom. 2017, 16, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guldbrandsen, A.; Vethe, H.; Farag, Y.; Oveland, E.; Garberg, H.; Berle, M.; Myhr, K.M.; Opsahl, J.A.; Barsnes, H.; Berven, F.S. In-depth Characterization of the Cerebrospinal Fluid (CSF) Proteome Displayed Through the CSF Proteome Resource (CSF-PR). Mol. Cell. Proteom. 2014, 13, 3152–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, N.W.; Conrads, T.P. Recent advances and opportunities in proteomic analyses of tumour heterogeneity. J. Pathol. 2018, 244, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omenn, G.S.; States, D.J.; Adamski, M.; Blackwell, T.W.; Menon, R.; Hermjakob, H.; Apweiler, R.; Haab, B.B.; Simpson, R.J.; Eddes, J.S.; et al. Overview of the HUPO Plasma Proteome Project: Results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 2005, 5, 3226–3245. [Google Scholar] [CrossRef]
- Rai, A.J.; Gelfand, C.A.; Haywood, B.C.; Warunek, D.J.; Yi, J.; Schuchard, M.D.; Mehigh, R.J.; Cockrill, S.L.; Scott, G.B.; Tammen, H.; et al. HUPO Plasma Proteome Project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Martinez-Val, A.; Steigerwald, S.; Ruther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.G.; Schilling, B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Rev. Proteom. 2017, 14, 419–429. [Google Scholar] [CrossRef]
- Domon, B.; Gallien, S. Recent advances in targeted proteomics for clinical applications. Proteom. Clin. Appl. 2015, 9, 423–431. [Google Scholar] [CrossRef]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A.; Niu, L.; Muller, J.B.; Buchholtz, M.L.; Bader, J.M.; Teupser, D.; et al. Plasma Proteome Profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Prognostic Factors | Reference(s) |
|---|---|
| Circulating blast cells at the time of diagnosis | [49] |
| Karyotype | [12] |
| Molecular genetics | [12] |
| Secondary AML | [50] |
| Metabolic status at the time of diagnosis | [51] |
| AML subclones at the time of diagnosis | [52,53,54] |
| No remaining blasts on light microscopy 14–17 days after start of induction chemotherapy | [55] |
| Complete hematological remission after first induction cycle | [49] |
| MRD after consolidation | [56,57] |
| Time of relapse: - After conventional therapy (i.e., duration of first complete remission) | [58,59] |
| - Time from ASCT until diagnosis of relapse | [60,61] |
| MS-Based Quantification | Sample Preparation Components | Clinical and Pathological Findings/Identified Marker(s) | Cohort Size a | Ref. |
|---|---|---|---|---|
| Label-free DDA | In-solution digestion, IP | Study on the protein tyrosine kinase-protein tyrosine phosphatase connections and effect on protein-phosphotyrosine signaling networks | 12 | [80] |
| Label-free DDA | FACS, in-solution digestion, high pH RP-fractionation | Altered expression of leukemia-enriched plasma membrane proteins on distinct AML subclones. Some of the proteins (e.g., IL3RA, TIM3, CD44, CD96, CD47, CD32, IL2RA, CD99, and CLEC12A) have been previously identified by other non-MS-based technologies | 42 | [81] |
| Label-free DDA | FASP | The constitutive release of mediators from primary AML differ from the intracellular protein levels | 19 | [82] |
| Label-free DDA, SRM | 2D-DIGE, IMAC | Phosphoprotein profiles reveal blast differentiation and cytogenic risk stratification | 62 | [83] |
| Label-free DDA | FASP | Patient subsets with high constitutive cytokine release levels show high expression of proteins involved in intracellular signaling interacting with integrins, RAC1, and SYK. AML cells with low release show high expression of transcriptional regulators | 16 | [84] |
| Label-free DDA | FASP | Strong antiproliferative and proapoptotic effects of metabolic pathways inhibitors on AML patient cells | 14 | [48] |
| Super-SILAC DDA | FASP, mixed-mode fractionation, IMAC | Higher phosphorylation of transcription regulators decreased cytokine release and increased integrin expression on cells from AML patients with high constitutive activation of the PI3K-AKT-mTOR signaling pathway | 20 | [85] |
| Super-SILAC DDA | FASP, mixed-mode fractionation, IMAC | Transcription factors and proteins involved in mRNA splicing are highly expressed in AML cells with self-renewal capacity | 15 | [86] |
| Super-SILAC DDA | FASP, mixed-mode fractionation, IMAC | Enhanced phosphorylation and activation of the PI3K-AKT-mTOR pathway by insulin is coupled to reduced antiproliferative effects of metabolic inhibitors in AML patient subsets | 14 | [68] |
| Super-SILAC DDA | FASP, mixed-mode fractionation, IMAC | High expression of RNA processing proteins, low expression of V-ATPase proteins, and higher activity of CSK2 and CDKs could help predict chemoresistant AML relapse | 41 | [46] |
| Super-SILAC DDA | FASP, mixed-mode fractionation, IMAC | High expression of mitochondrial ribosomal subunit proteins, RNA processing proteins, DNA repair proteins, and high activity of CDKs at AML relapse | 14 b | [47] |
| TMT DDA | FACS, LSCs engraftment, in-solution digestion, high pH RP-fractionation | Characterization of the expression of cell adhesion molecules, proteins of the oxidative phosphorylation process, and spliceosome factors in LSCs | 18 c | [87] |
| TMT DDA | FACS, LSCs engraftment, in-solution digestion, high pH RP-fractionation | BCAT1 is enriched in LSCs and links BCAA metabolism to epigenomic and post-translational HIF1α regulation via αKG-dependent dioxygenase | 18 c | [88] |
| TMT DDA | FACS, membrane, and cytosol isolation, in-solution digestion | Protein modification and cytoskeleton reorganization proteins showed an altered abundance in the proteome of leukemic progenitor cells | 5 | [89] |
| iTRAQ DDA | Nuclear isolation, in-solution digestion, high-pH RP-fractionation | Over-expression of nuclear S100A4 in AML cells. Nuclear S100A4 is crucial for AML survival | 15 | [90] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Valladares, M.; Bruserud, Ø.; Selheim, F. The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives. Int. J. Mol. Sci. 2020, 21, 6830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186830
Hernandez-Valladares M, Bruserud Ø, Selheim F. The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives. International Journal of Molecular Sciences. 2020; 21(18):6830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186830
Chicago/Turabian StyleHernandez-Valladares, Maria, Øystein Bruserud, and Frode Selheim. 2020. "The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives" International Journal of Molecular Sciences 21, no. 18: 6830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186830