
New Therapeutics Targeting Arterial Media Calcification: Friend or Foe for Bone Mineralization?




{kind=link}
Abstract
:1. Introduction
2. An Overview of Pivotal Cellular and Molecular Mechanisms of Artery and Bone Mineralization
2.1. Targeting the High Phenotypic Plasticity in the Vasculature
2.2. Targeting Circulating Calcification Inhibitors and Stimulators
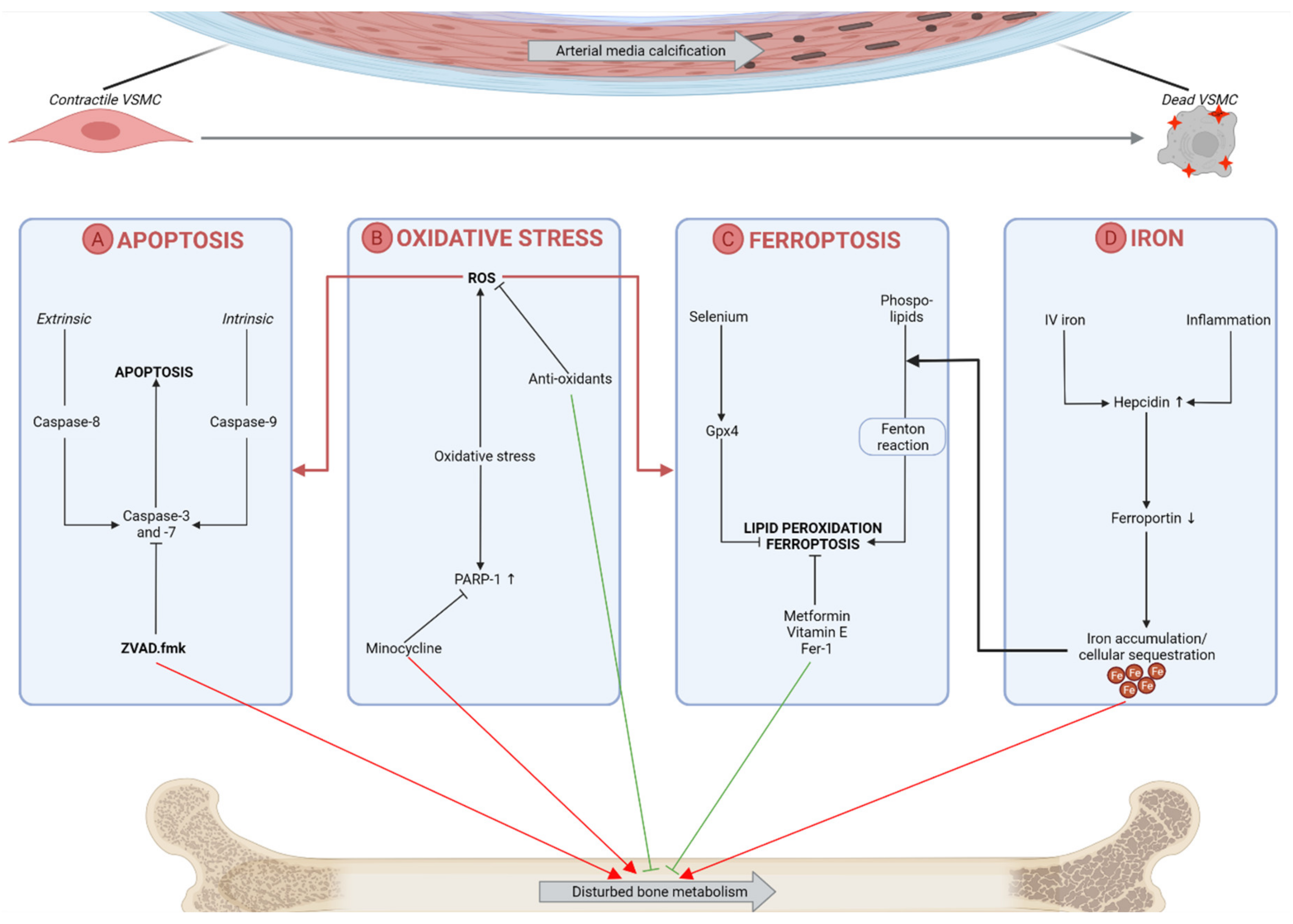
2.3. Targeting Cell Death Events in the Vasculature
2.4. Targeting Oxidative Stress in the Vasculature
3. Nutritional Care to Treat Arterial Media Calcification
4. Towards a Local Approach to Tackle Arterial Media Calcification
5. Extrapolation of Anti-Arterial Media Calcification Therapeutics toward the Human Situation and Other Types of Cardiovascular Calcification
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nigwekar, S.U.; Thadhani, R.; Brandenburg, V.M. Calciphylaxis. New Engl. J. Med. 2018, 378, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Aikawa, E.; Merryman, W.D. Potential drug targets for calcific aortic valve disease. Nat. Rev. Cardiol. 2014, 11, 218–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, E.J.; Nicholls, S.J.; Bartolo, B.A.D. Plaque Calcification. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Tölle, M.; Reshetnik, A.; Schuchardt, M.; Höhne, M.; van der Giet, M. Arteriosclerosis and vascular calcification: Causes, clinical assessment and therapy. Eur. J. Clin. Investig. 2015, 45, 976–985. [Google Scholar] [CrossRef]
- De Vilder, E.Y.; Vanakker, O.M. From variome to phenome: Pathogenesis, diagnosis and management of ectopic mineralization disorders. World J. Clin. Cases 2015, 3, 556–574. [Google Scholar] [CrossRef]
- Cho, I.J.; Chang, H.J.; Park, H.B.; Heo, R.; Shin, S.; Shim, C.Y.; Hong, G.R.; Chung, N. Aortic calcification is associated with arterial stiffening, left ventricular hypertrophy, and diastolic dysfunction in elderly male patients with hypertension. J. Hypertens. 2015, 33, 1633–1641. [Google Scholar] [CrossRef]
- Sigrist, M.K.; Taal, M.W.; Bungay, P.; McIntyre, C.W. Progressive Vascular Calcification over 2 Years Is Associated with Arterial Stiffening and Increased Mortality in Patients with Stages 4 and 5 Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 1241. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-R.; Zhang, J.-J.; Xu, X.-X.; Wu, Y.-G. Prevalence of coronary artery calcification and its association with mortality, cardiovascular events in patients with chronic kidney disease: A systematic review and meta-analysis. Ren. Fail. 2019, 41, 244–256. [Google Scholar] [CrossRef]
- Porter, C.J.; Stavroulopoulos, A.; Roe, S.D.; Pointon, K.; Cassidy, M.J.D. Detection of coronary and peripheral artery calcification in patients with chronic kidney disease stages 3 and 4, with and without diabetes. Nephrol. Dial. Transplant. 2007, 22, 3208–3213. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56-e528. [Google Scholar] [CrossRef]
- Evenepoel, P.; Opdebeeck, B.; David, K.; D’Haese, P.C. Bone-Vascular Axis in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 472–483. [Google Scholar] [CrossRef]
- Singh, A.; Tandon, S.; Tandon, C. An update on vascular calcification and potential therapeutics. Mol. Biol. Rep. 2021, 48, 887–896. [Google Scholar] [CrossRef]
- Neven, E.; De Schutter, T.M.; De Broe, M.E.; D’Haese, P.C. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011, 79, 1166–1177. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Neven, E.; Dauwe, S.; De Broe, M.E.; D’Haese, P.C.; Persy, V. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int. 2007, 72, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, P.; Chen, N.X.; O’Neill, K.; McClintick, J.N.; Moe, S.M.; Janga, S.C. Differential miRNA Expression in Cells and Matrix Vesicles in Vascular Smooth Muscle Cells from Rats with Kidney Disease. PLoS ONE 2015, 10, e0131589. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Feng, W.; Su, X.; Luo, D.; Li, Z.; Zhou, Y.; Zhu, Y.; Zhang, M.; Chen, J.; Liu, B.; et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J. Clin. Investig. 2022, 132, e150051. [Google Scholar] [CrossRef]
- Schelski, N.; Luong, T.T.D.; Lang, F.; Pieske, B.; Voelkl, J.; Alesutan, I. SGK1-dependent stimulation of vascular smooth muscle cell osteo-/chondrogenic transdifferentiation by interleukin-18. Pflug. Arch. Eur. J. Physiol. 2019, 471, 889–899. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Xu, J.; Bai, Y.; Zhang, H.; Zhou, W.; Cheng, M.; Zhang, D.; Zhang, L.; Zhang, S. MicroRNA-103a regulates the calcification of vascular smooth muscle cells by targeting runt-related transcription factor 2 in high phosphorus conditions. Exp. Ther. Med. 2021, 22, 1036. [Google Scholar] [CrossRef]
- Wang, L.; Chennupati, R.; Jin, Y.J.; Li, R.; Wang, S.; Günther, S.; Offermanns, S. YAP/TAZ Are Required to Suppress Osteogenic Differentiation of Vascular Smooth Muscle Cells. iScience 2020, 23, 101860. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019, 25, 1133–1146. [Google Scholar] [CrossRef]
- De Mare, A.; Maudsley, S.; Azmi, A.; Hendrickx, J.O.; Opdebeeck, B.; Neven, E.; D’Haese, P.C.; Verhulst, A. Sclerostin as Regulatory Molecule in Vascular Media Calcification and the Bone-Vascular Axis. Toxins 2019, 11, 428. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Bennett, C.N.; Gerin, I.; Rapp, L.A.; Hankenson, K.D.; Macdougald, O.A. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 14515–14524. [Google Scholar] [CrossRef]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Zhou, Y.; Liu, Y.; Xie, M.; Guo, G. Inhibitory Effect of Sirtuin6 (SIRT6) on Osteogenic Differentiation of Bone Marrow Mesenchymal Stem Cells. Med. Sci. Monit. 2019, 25, 8412–8421. [Google Scholar] [CrossRef]
- Zuo, B.; Zhu, J.; Li, J.; Wang, C.; Zhao, X.; Cai, G.; Li, Z.; Peng, J.; Wang, P.; Shen, C.; et al. microRNA-103a functions as a mechanosensitive microRNA to inhibit bone formation through targeting Runx2. J. Bone Miner. Res. 2015, 30, 330–345. [Google Scholar] [CrossRef]
- Patel, J.J.; Bourne, L.E.; Davies, B.K.; Arnett, T.R.; MacRae, V.E.; Wheeler-Jones, C.P.; Orriss, I.R. Differing calcification processes in cultured vascular smooth muscle cells and osteoblasts. Exp. Cell Res. 2019, 380, 100–113. [Google Scholar] [CrossRef]
- Medici, D.; Kalluri, R. Endothelial–mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Tang, R.-N.; Liu, H.; Pan, M.-M.; Liu, B.-C. Cinacalcet ameliorates aortic calcification in uremic rats via suppression of endothelial-to-mesenchymal transition. Acta Pharmacol. Sin. 2016, 37, 1423–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasuri, F.; Valente, S.; Motta, I.; Degiovanni, A.; Ciavarella, C.; Pasquinelli, G. ETS-Related Gene Expression in Healthy Femoral Arteries with Focal Calcifications. Front. Cell Dev. Biol. 2021, 9, 623782. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.-J.; ten Dijke, P. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes Snail-mediated endothelial–mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 Regulates Osterix through Msx2 and Runx2 during Osteoblast Differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; Schneider, R.K.; Kuppe, C.; Kaesler, N.; Chang-Panesso, M.; Machado, F.G.; Gratwohl, S.; et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem Cell 2016, 19, 628–642. [Google Scholar] [CrossRef] [Green Version]
- Boström, K.; Watson, K.E.; Horn, S.; Wortham, C.; Herman, I.M.; Demer, L.L. Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Investig. 1993, 91, 1800–1809. [Google Scholar] [CrossRef]
- Andreeva, E.R.; Pugach, I.M.; Gordon, D.; Orekhov, A.N. Continuous subendothelial network formed by pericyte-like cells in human vascular bed. Tissue Cell 1998, 30, 127–135. [Google Scholar] [CrossRef]
- Farrington-Rock, C.; Crofts, N.J.; Doherty, M.J.; Ashton, B.A.; Griffin-Jones, C.; Canfield, A.E. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation 2004, 110, 2226–2232. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Flores, L.; Gutierrez, R.; Lopez-Alonso, A.; Gonzalez, R.; Varela, H. Pericytes as a supplementary source of osteoblasts in periosteal osteogenesis. Clin. Orthop. Relat. Res. 1992, 275, 280–286. [Google Scholar] [CrossRef]
- Thomas, W.E. Brain macrophages: On the role of pericytes and perivascular cells. Brain Res. Rev. 1999, 31, 42–57. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Smyth, L.C.; Jansson, D.; Schweder, P.; Aalderink, M.; Scotter, E.L.; Mee, E.W.; Faull, R.L.M.; Park, T.I.; Dragunow, M. Modelling physiological and pathological conditions to study pericyte biology in brain function and dysfunction. BMC Neurosci. 2018, 19, 6. [Google Scholar] [CrossRef]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement from the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef]
- Eckardt, K.-U.; Kasiske, B.L. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 76, S1–S130. [Google Scholar] [CrossRef]
- Fleisch, H.; Russell, R.G.; Straumann, F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 1966, 212, 901–903. [Google Scholar] [CrossRef]
- Dedinszki, D.; Szeri, F.; Kozák, E.; Pomozi, V.; Tőkési, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Orriss, I.R.; Neven, E.; D’Haese, P.C.; Verhulst, A. Extracellular Nucleotides Regulate Arterial Calcification by Activating Both Independent and Dependent Purinergic Receptor Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 7636. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone 2020, 137, 115392. [Google Scholar] [CrossRef]
- Patel, J.J.; Zhu, D.; Opdebeeck, B.; D’Haese, P.; Millán, J.L.; Bourne, L.E.; Wheeler-Jones, C.P.D.; Arnett, T.R.; MacRae, V.E.; Orriss, I.R. Inhibition of arterial medial calcification and bone mineralization by extracellular nucleotides: The same functional effect mediated by different cellular mechanisms. J. Cell. Physiol. 2018, 233, 3230–3243. [Google Scholar] [CrossRef]
- Allen, M.R. Preclinical Models for Skeletal Research: How Commonly Used Species Mimic (or Don’t) Aspects of Human Bone. Toxicol. Pathol. 2017, 45, 851–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme. Pharmaceutics 2021, 13, 1138. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Wang, X.; Millán, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol.—Heart Circ. Physiol. 2011, 301, H61–H68. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, A.; Eckert, T.; Aretz, A.; Richtering, W.; van Dorp, W.; Schäfer, C.; Jahnen-Dechent, W. Hierarchical Role of Fetuin-A and Acidic Serum Proteins in the Formation and Stabilization of Calcium Phosphate Particles. J. Biol. Chem. 2008, 283, 14815–14825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Böhm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Perelló, J.; Joubert, P.H.; Ferrer, M.D.; Canals, A.Z.; Sinha, S.; Salcedo, C. First-time-in-human randomized clinical trial in healthy volunteers and haemodialysis patients with SNF472, a novel inhibitor of vascular calcification. Br. J. Clin. Pharmacol. 2018, 84, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, C.; Joubert, P.H.; Ferrer, M.D.; Canals, A.Z.; Maduell, F.; Torregrosa, V.; Campistol, J.M.; Ojeda, R.; Perelló, J. A phase 1b randomized, placebo-controlled clinical trial with SNF472 in haemodialysis patients. Br. J. Clin. Pharmacol. 2019, 85, 796–806. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Bellasi, A.; Bushinsky, D.; Bover, J.; Rodriguez, M.; Ketteler, M.; Sinha, S.; Salcedo, C.; Gillotti, K.; Padgett, C.; et al. Slowing Progression of Cardiovascular Calcification with SNF472 in Patients on Hemodialysis. Circulation 2020, 141, 728–739. [Google Scholar] [CrossRef]
- Sinha, S.; Gould, L.J.; Nigwekar, S.U.; Serena, T.E.; Brandenburg, V.; Moe, S.M.; Aronoff, G.; Chatoth, D.K.; Hymes, J.L.; Miller, S.; et al. The CALCIPHYX study: A randomized, double-blind, placebo-controlled, Phase 3 clinical trial of SNF472 for the treatment of calciphylaxis. Clin. Kidney J. 2021, 15, 136–144. [Google Scholar] [CrossRef]
- Perelló, J.; Ferrer, M.D.; del Mar Pérez, M.; Kaesler, N.; Brandenburg, V.M.; Behets, G.J.; D’Haese, P.C.; Garg, R.; Isern, B.; Gold, A.; et al. Mechanism of action of SNF472, a novel calcification inhibitor to treat vascular calcification and calciphylaxis. Br. J. Pharmacol. 2020, 177, 4400–4415. [Google Scholar] [CrossRef]
- Schantl, A.E.; Verhulst, A.; Neven, E.; Behets, G.J.; D’Haese, P.C.; Maillard, M.; Mordasini, D.; Phan, O.; Burnier, M.; Spaggiari, D.; et al. Inhibition of vascular calcification by inositol phosphates derivatized with ethylene glycol oligomers. Nat. Commun. 2020, 11, 721. [Google Scholar] [CrossRef]
- Bushinsky, D.A.; Raggi, P.; Bover, J.; Ketteler, M.; Bellasi, A.; Rodriguez, M.; Sinha, S.; Garg, R.; Perelló, J.; Gold, A.; et al. Effects of Myo-inositol Hexaphosphate (SNF472) on Bone Mineral Density in Patients Receiving Hemodialysis. Clin. J. Am. Soc. Nephrol. 2021, 16, 736–745. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef]
- Danziger, J. Vitamin K-dependent proteins, warfarin, and vascular calcification. Clin. J. Am. Soc. Nephrol. 2008, 3, 1504–1510. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Schoppet, M.; Schurgers, L.J.; Reynolds, J.L.; McNair, R.; Heiss, A.; Jahnen-Dechent, W.; Hackeng, T.M.; Schlieper, G.; Harrison, P.; et al. Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arterioscler. Thromb. Vasc. 2017, 37, e22–e32. [Google Scholar] [CrossRef] [Green Version]
- Cranenburg, E.C.; Schurgers, L.J.; Uiterwijk, H.H.; Beulens, J.W.; Dalmeijer, G.W.; Westerhuis, R.; Magdeleyns, E.J.; Herfs, M.; Vermeer, C.; Laverman, G.D. Vitamin K intake and status are low in hemodialysis patients. Kidney Int. 2012, 82, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Holden, R.M.; Morton, A.R.; Garland, J.S.; Pavlov, A.; Day, A.G.; Booth, S.L. Vitamins K and D status in stages 3–5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 590–597. [Google Scholar] [CrossRef]
- Malhotra, R.; Burke, M.F.; Martyn, T.; Shakartzi, H.R.; Thayer, T.E.; O’Rourke, C.; Li, P.; Derwall, M.; Spagnolli, E.; Kolodziej, S.A.; et al. Inhibition of Bone Morphogenetic Protein Signal Transduction Prevents the Medial Vascular Calcification Associated with Matrix Gla Protein Deficiency. PLoS ONE 2015, 10, e0117098. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Mare, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Caluwé, R.; Pyfferoen, L.; De Bacquer, D.; De Boeck, K.; Delanote, J.; De Surgeloose, D.; Van Hoenacker, P.; Van Vlem, B.; Verbeke, F. Multicenter Randomized Controlled Trial of Vitamin K Antagonist Replacement by Rivaroxaban with or without Vitamin K2 in Hemodialysis Patients with Atrial Fibrillation: The Valkyrie Study. J. Am. Soc. Nephrol. 2019, 31, 186–196. [Google Scholar] [CrossRef]
- Grzejszczak, P.; Kurnatowska, I. Role of Vitamin K in CKD: Is Its Supplementation Advisable in CKD Patients? Kidney Blood Press. Res. 2021, 46, 523–530. [Google Scholar] [CrossRef]
- Zwakenberg, S.R.; de Jong, P.A.; Bartstra, J.W.; van Asperen, R.; Westerink, J.; de Valk, H.; Slart, R.H.J.A.; Luurtsema, G.; Wolterink, J.M.; de Borst, G.J.; et al. The effect of menaquinone-7 supplementation on vascular calcification in patients with diabetes: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2019, 110, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Bartstra, J.W.; Draaisma, F.; Zwakenberg, S.R.; Lessmann, N.; Wolterink, J.M.; van der Schouw, Y.T.; de Jong, P.A.; Beulens, J.W.J. Six months vitamin K treatment does not affect systemic arterial calcification or bone mineral density in diabetes mellitus 2. Eur. J. Nutr. 2021, 60, 1691–1699. [Google Scholar] [CrossRef]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [Green Version]
- Cazaña-Pérez, V.; Cidad, P.; Donate-Correa, J.; Martín-Núñez, E.; López-López, J.R.; Pérez-García, M.T.; Giraldez, T.; Navarro-González, J.F.; Alvarez de la Rosa, D. Phenotypic Modulation of Cultured Primary Human Aortic Vascular Smooth Muscle Cells by Uremic Serum. Front. Physiol. 2018, 9, 89. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Farzaneh-Far, A.; Shanahan, C.M.; Weissberg, P.L. The role of apoptosis in the initiation of vascular calcification. Z. Kardiol. 2001, 90, 43–46. [Google Scholar] [CrossRef]
- Coscas, R.; Bensussan, M.; Jacob, M.-P.; Louedec, L.; Massy, Z.; Sadoine, J.; Daudon, M.; Chaussain, C.; Bazin, D.; Michel, J.-B. Free DNA precipitates calcium phosphate apatite crystals in the arterial wall in vivo. Atherosclerosis 2017, 259, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Reedy, M.C.; Hannun, Y.A.; Obeid, L.M. Inhibition of Caspases Inhibits the Release of Apoptotic Bodies: Bcl-2 Inhibits the Initiation of Formation of Apoptotic Bodies in Chemotherapeutic Agent-induced Apoptosis. J. Cell Biol. 1999, 145, 99–108. [Google Scholar] [CrossRef]
- Adamova, E.; Janeckova, E.; Kleparnik, K.; Matalova, E. Caspases and osteogenic markers—In vitro screening of inhibition impact. In Vitro Cell Dev. Biol 2016, 52, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A. Activation of caspases is required for osteoblastic differentiation. J. Biol. Chem. 2003, 278, 47477–47482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janečková, E.; Bíliková, P.; Matalová, E. Osteogenic Potential of Caspases Related to Endochondral Ossification. J. Histochem. Cytochem. 2017, 66, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; McGill, M.R.; Chao, X.; Woolbright, B.L.; Jaeschke, H.; Ding, W.-X. Caspase Inhibition Prevents Tumor Necrosis Factor-α–Induced Apoptosis and Promotes Necrotic Cell Death in Mouse Hepatocytes in Vivo and in Vitro. Am. J. Pathol. 2016, 186, 2623–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaire, C.; Andréau, K.; Souvannavong, V.; Adam, A. Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett. 1998, 425, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef]
- Kawtharany, L.; Bessueille, L.; Issa, H.; Hamade, E.; Zibara, K.; Magne, D. Inflammation and Microcalcification: A Never-Ending Vicious Cycle in Atherosclerosis? J. Vasc. Res. 2022, 1–14. [Google Scholar] [CrossRef]
- Canet-Soulas, E.; Bessueille, L.; Mechtouff, L.; Magne, D. The Elusive Origin of Atherosclerotic Plaque Calcification. Front. Cell Dev. Biol. 2021, 9, 622736. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Do TUNEL and Other Apoptosis Assays Detect Cell Death in Preclinical Studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Zhang, S.; Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 2021, 11, 3052–3059. [Google Scholar] [CrossRef]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef]
- You, H.; Yang, H.; Zhu, Q.; Li, M.; Xue, J.; Gu, Y.; Lin, S.; Ding, F. Advanced oxidation protein products induce vascular calcification by promoting osteoblastic trans-differentiation of smooth muscle cells via oxidative stress and ERK pathway. Ren. Fail. 2009, 31, 313–319. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Qin, F.; Huang, K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J. Biol. Inorg. Chem. 2014, 19, 375–388. [Google Scholar] [CrossRef]
- Nakanishi, T.; Hasuike, Y.; Otaki, Y.; Kida, A.; Nonoguchi, H.; Kuragano, T. Hepcidin: Another culprit for complications in patients with chronic kidney disease? Nephrol. Dial. Transplant. 2011, 26, 3092–3100. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.Y.; Zhang, M.; Chen, S.L.; Zhang, S.P.; Guo, C.Y.; Wang, J.S.; Liu, X.; Miao, Y.; Yin, H.J. The Influence of Hyperlipidemia on Endothelial Function of FPN1 Tek-Cre Mice and the Intervention Effect of Tetramethylpyrazine. Cell. Physiol. Biochem. 2018, 47, 119–128. [Google Scholar] [CrossRef]
- Kawada, S.; Nagasawa, Y.; Kawabe, M.; Ohyama, H.; Kida, A.; Kato-Kogoe, N.; Nanami, M.; Hasuike, Y.; Kuragano, T.; Kishimoto, H.; et al. Iron-induced calcification in human aortic vascular smooth muscle cells through interleukin-24 (IL-24), with/without TNF-alpha. Sci. Rep. 2018, 8, 658. [Google Scholar] [CrossRef]
- Wong, S.K.; Mohamad, N.-V.; Ibrahim, N.I.; Chin, K.-Y.; Shuid, A.N.; Ima-Nirwana, S. The Molecular Mechanism of Vitamin E as a Bone-Protecting Agent: A Review on Current Evidence. Int. J. Mol. Sci. 2019, 20, 1453. [Google Scholar] [CrossRef] [Green Version]
- Valanezhad, A.; Odatsu, T.; Abe, S.; Watanabe, I. Bone Formation Ability and Cell Viability Enhancement of MC3T3-E1 Cells by Ferrostatin-1 a Ferroptosis Inhibitor of Cancer Cells. Int. J. Mol. Sci. 2021, 22, 12259. [Google Scholar] [CrossRef]
- Ma, W.-Q.; Sun, X.-J.; Zhu, Y.; Liu, N.-F. Metformin attenuates hyperlipidaemia-associated vascular calcification through anti-ferroptotic effects. Free Radic. Biol. Med. 2021, 165, 229–242. [Google Scholar] [CrossRef]
- Cortizo, A.M.; Sedlinsky, C.; McCarthy, A.D.; Blanco, A.; Schurman, L. Osteogenic actions of the anti-diabetic drug metformin on osteoblasts in culture. Eur. J. Pharmacol. 2006, 536, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Jiating, L.; Buyun, J.; Yinchang, Z. Role of Metformin on Osteoblast Differentiation in Type 2 Diabetes. BioMed Res. Int. 2019, 2019, 9203934. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid. Med. Cell. Longev. 2020, 2020, 9067610. [Google Scholar] [CrossRef]
- Jin, D.; Lin, L.; Xie, Y.; Jia, M.; Qiu, H.; Xun, K. NRF2-suppressed vascular calcification by regulating the antioxidant pathway in chronic kidney disease. FASEB J. 2022, 36, e22098. [Google Scholar] [CrossRef]
- Chen, W.R.; Zhou, Y.J.; Yang, J.Q.; Liu, F.; Wu, X.P.; Sha, Y. Melatonin Attenuates Calcium Deposition from Vascular Smooth Muscle Cells by Activating Mitochondrial Fusion and Mitophagy via an AMPK/OPA1 Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 5298483. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Wang, Z.; Li, C.; Li, S.; Li, L.; Fan, Q.; Zheng, L. Melatonin Suppresses Estrogen Deficiency-Induced Osteoporosis and Promotes Osteoblastogenesis by Inactivating the NLRP3 Inflammasome. Calcif. Tissue Int. 2018, 103, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Da, W.; Tao, L.; Wen, K.; Tao, Z.; Wang, S.; Zhu, Y. Protective Role of Melatonin Against Postmenopausal Bone Loss via Enhancement of Citrate Secretion from Osteoblasts. Front. Pharmacol. 2020, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.J.; Tsai, H.-C.; Chang, A.-C.; Huang, C.-C.; Yang, S.-F.; Tang, C.-H. Melatonin Inhibits Osteoclastogenesis and Osteolytic Bone Metastasis: Implications for Osteoporosis. Int. J. Mol. Sci. 2021, 22, 9435. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Hagiwara, H. Excess iron inhibits osteoblast metabolism. Toxicol. Lett. 2009, 191, 211–215. [Google Scholar] [CrossRef]
- Jeney, V. Clinical Impact and Cellular Mechanisms of Iron Overload-Associated Bone Loss. Front. Pharmacol. 2017, 8, 77. [Google Scholar] [CrossRef]
- Hou, J.-M.; Xue, Y.; Lin, Q.-M. Bovine lactoferrin improves bone mass and microstructure in ovariectomized rats via OPG/RANKL/RANK pathway. Acta Pharmacol. Sin. 2012, 33, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Seto, T.; Hamada, C.; Tomino, Y. Suppressive effects of iron overloading on vascular calcification in uremic rats. J. Nephrol. 2014, 27, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Neven, E.; Corremans, R.; Vervaet, B.A.; Funk, F.; Walpen, S.; Behets, G.J.; D’Haese, P.C.; Verhulst, A. Renoprotective effects of sucroferric oxyhydroxide in a rat model of chronic renal failure. Nephrol. Dial. Transplant. 2020, 35, 1689–1699. [Google Scholar] [CrossRef]
- Tóth, A.; Balogh, E.; Jeney, V. Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants 2020, 9, 963. [Google Scholar] [CrossRef]
- Boraldi, F.; Lofaro, F.D.; Quaglino, D. Apoptosis in the Extraosseous Calcification Process. Cells 2021, 10, 131. [Google Scholar] [CrossRef]
- Agidigbi, T.S.; Kim, C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int. J. Mol. Sci. 2019, 20, 3576. [Google Scholar] [CrossRef] [Green Version]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216. [Google Scholar] [CrossRef]
- Chang, X.-Y.; Cui, L.; Wang, X.-Z.; Zhang, L.; Zhu, D.; Zhou, X.-R.; Hao, L.-R. Quercetin Attenuates Vascular Calcification through Suppressed Oxidative Stress in Adenine-Induced Chronic Renal Failure Rats. BioMed Res. Int. 2017, 2017, 5716204. [Google Scholar] [CrossRef]
- Manivannan, J.; Barathkumar, T.R.; Sivasubramanian, J.; Arunagiri, P.; Raja, B.; Balamurugan, E. Diosgenin attenuates vascular calcification in chronic renal failure rats. Mol. Cell. Biochem. 2013, 378, 9–18. [Google Scholar] [CrossRef]
- Ji, R.; Sun, H.; Peng, J.; Ma, X.; Bao, L.; Fu, Y.; Zhang, X.; Luo, C.; Gao, C.; Jin, Y.; et al. Rosmarinic acid exerts an antagonistic effect on vascular calcification by regulating the Nrf2 signalling pathway. Free Radic. Res. 2019, 53, 187–197. [Google Scholar] [CrossRef]
- Pasch, A.; Schaffner, T.; Huynh-Do, U.; Frey, B.M.; Frey, F.J.; Farese, S. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008, 74, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Liu, F.; Dai, X.; Zhou, L.; Fu, P. Sodium thiosulfate protects human aortic smooth muscle cells from osteoblastic transdifferentiation via high-level phosphate. Kaohsiung J. Med. Sci. 2013, 29, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.-T.; Yeh, H.-Y.; Tsai, Y.-T.; Chuang, P.-H.; Yuan, T.-H.; Huang, J.-W.; Chen, H.-W. Natural and non-natural antioxidative compounds: Potential candidates for treatment of vascular calcification. Cell Death Discov. 2019, 5, 145. [Google Scholar] [CrossRef]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: Important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef]
- Bassi, E.; Liberman, M.; Martinatti, M.K.; Bortolotto, L.A.; Laurindo, F.R.M. Lipoic acid, but not tempol, preserves vascular compliance and decreases medial calcification in a model of elastocalcinosis. Braz. J. Med. Biol. Res. 2014, 47, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, W.; An, J.; Liang, M.; Li, Y.; Zhang, F.; Tong, Q.; Huang, K. Poly(ADP-ribose) polymerase 1 accelerates vascular calcification by upregulating Runx2. Nat. Commun. 2019, 10, 1203. [Google Scholar] [CrossRef] [Green Version]
- Muller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, Y.; Fujihara, H.; Kawaguchi, K.; Yamada, H.; Nakayama, R.; Yamamoto, N.; Fujihara, Y.; Hamada, Y.; Satomura, K.; Masutani, M. PARP Inhibitor PJ34 Suppresses Osteogenic Differentiation in Mouse Mesenchymal Stem Cells by Modulating BMP-2 Signaling Pathway. Int. J. Mol. Sci. 2015, 16, 24820–24838. [Google Scholar] [CrossRef] [PubMed]
- Beazley, K.E.; Lima, F.; Borras, T.; Nurminskaya, M. Attenuation of chondrogenic transformation in vascular smooth muscle by dietary quercetin in the MGP-deficient mouse model. PLoS ONE 2013, 8, e76210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vasc. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef]
- Hou, M.; Song, Y.; Li, Z.; Luo, C.; Ou, J.S.; Yu, H.; Yan, J.; Lu, L. Curcumin attenuates osteogenic differentiation and calcification of rat vascular smooth muscle cells. Mol. Cell. Biochem. 2016, 420, 151–160. [Google Scholar] [CrossRef]
- Mehansho, H.; Majeti, S.; Tzeghai, G. Prevention of Vascular Calcification by Magnesium and Selected Polyphenols. Adv. Prev. Med. 2021, 2021, 6686597. [Google Scholar] [CrossRef]
- Shioi, A.; Morioka, T.; Shoji, T.; Emoto, M. The Inhibitory Roles of Vitamin K in Progression of Vascular Calcification. Nutrients 2020, 12, 583. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Raggi, P.; Chertow, G.M. SNF472: Mechanism of action and results from clinical trials. Curr. Opin. Nephrol. Hypertens. 2021, 30, 424–429. [Google Scholar] [CrossRef]
- Salminen, W.; Agbaje-Williams, M.; Ajayi, F.O. A Unique Formulation of Cardioprotective Bio-Actives: An Overview of Their Safety Profile. Medicines 2019, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Koshihara, Y.; Hoshi, K.; Okawara, R.; Ishibashi, H.; Yamamoto, S. Vitamin K stimulates osteoblastogenesis and inhibits osteoclastogenesis in human bone marrow cell culture. J. Endocrinol. 2003, 176, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, S.; Liu, J.; Liu, Y.; Liang, Q. Vitamin K2 stimulates MC3T3-E1 osteoblast differentiation and mineralization through autophagy induction. Mol. Med. Rep. 2019, 19, 3676–3684. [Google Scholar] [CrossRef]
- Ter Braake, A.D.; Shanahan, C.M.; De Baaij, J.H.F. Magnesium Counteracts Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar] [CrossRef] [Green Version]
- Kircelli, F.; Peter, M.E.; Sevinc Ok, E.; Celenk, F.G.; Yilmaz, M.; Steppan, S.; Asci, G.; Ok, E.; Passlick-Deetjen, J. Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol. Dial. Transplant. 2011, 27, 514–521. [Google Scholar] [CrossRef]
- Diaz-Tocados, J.M.; Peralta-Ramirez, A.; Rodríguez-Ortiz, M.E.; Raya, A.I.; Lopez, I.; Pineda, C.; Herencia, C.; de Oca, A.M.; Vergara, N.; Steppan, S.; et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int. 2017, 92, 1084–1099. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Obi, Y.; Monden, C.; Oka, T.; Yamaguchi, S.; Matsui, I.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J. Am. Soc. Nephrol. 2019, 30, 1073–1085. [Google Scholar] [CrossRef]
- Tzanakis, I.P.; Stamataki, E.E.; Papadaki, A.N.; Giannakis, N.; Damianakis, N.E.; Oreopoulos, D.G. Magnesium retards the progress of the arterial calcifications in hemodialysis patients: A pilot study. Int. Urol. Nephrol. 2014, 46, 2199–2205. [Google Scholar] [CrossRef]
- Groenendijk, I.; van Delft, M.; Versloot, P.; van Loon, L.J.C.; de Groot, L.C.P.G.M. Impact of magnesium on bone health in older adults: A systematic review and meta-analysis. Bone 2022, 154, 116233. [Google Scholar] [CrossRef]
- Abedin, M.; Lim, J.; Tang, T.B.; Park, D.; Demer, L.L.; Tintut, Y. N-3 fatty acids inhibit vascular calcification via the p38-mitogen-activated protein kinase and peroxisome proliferator-activated receptor-gamma pathways. Circ. Res. 2006, 98, 727–729. [Google Scholar] [CrossRef]
- Kanai, S.; Uto, K.; Honda, K.; Hagiwara, N.; Oda, H. Eicosapentaenoic acid reduces warfarin-induced arterial calcification in rats. Atherosclerosis 2011, 215, 43–51. [Google Scholar] [CrossRef]
- Saito, Y.; Nakamura, K.; Miura, D.; Yunoki, K.; Miyoshi, T.; Yoshida, M.; Kawakita, N.; Kimura, T.; Kondo, M.; Sarashina, T.; et al. Suppression of Wnt Signaling and Osteogenic Changes in Vascular Smooth Muscle Cells by Eicosapentaenoic Acid. Nutrients 2017, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Sharma, T.; Mandal, C.C. Omega-3 fatty acids in pathological calcification and bone health. J. Food Biochem. 2020, 44, e13333. [Google Scholar] [CrossRef]
- Sun, D.; Krishnan, A.; Zaman, K.; Lawrence, R.; Bhattacharya, A.; Fernandes, G. Dietary n-3 Fatty Acids Decrease Osteoclastogenesis and Loss of Bone Mass in Ovariectomized Mice. J. Bone Miner. Res. 2003, 18, 1206–1216. [Google Scholar] [CrossRef]
- Raynor, W.Y.; Park, P.S.U.; Borja, A.J.; Sun, Y.; Werner, T.J.; Ng, S.J.; Lau, H.C.; Høilund-Carlsen, P.F.; Alavi, A.; Revheim, M.-E. PET-Based Imaging with 18F-FDG and 18F-NaF to Assess Inflammation and Microcalcification in Atherosclerosis and Other Vascular and Thrombotic Disorders. Diagnostics 2021, 11, 2234. [Google Scholar] [CrossRef]
- Sinha, A.; Shaporev, A.; Nosoudi, N.; Lei, Y.; Vertegel, A.; Lessner, S.; Vyavahare, N. Nanoparticle targeting to diseased vasculature for imaging and therapy. Nanomedicine 2014, 10, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Pai, A.S.; Giachelli, C.M. Matrix Remodeling in Vascular Calcification Associated with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2010, 21, 1637–1640. [Google Scholar] [CrossRef]
- Dao, H.H.; Essalihi, R.; Bouvet, C.; Moreau, P. Evolution and modulation of age-related medial elastocalcinosis: Impact on large artery stiffness and isolated systolic hypertension. Cardiovasc. Res. 2005, 66, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Karamched, S.R.; Nosoudi, N.; Moreland, H.E.; Chowdhury, A.; Vyavahare, N.R. Site-specific chelation therapy with EDTA-loaded albumin nanoparticles reverses arterial calcification in a rat model of chronic kidney disease. Sci. Rep. 2019, 9, 2629. [Google Scholar] [CrossRef]
- Dhital, S.; Rice, C.D.; Vyavahare, N.R. Reversal of elastase-induced abdominal aortic aneurysm following the delivery of nanoparticle-based pentagalloyl glucose (PGG) is associated with reduced inflammatory and immune markers. Eur. J. Pharmacol. 2021, 910, 174487. [Google Scholar] [CrossRef]
- Dhital, S.; Vyavahare, N.R. Nanoparticle-based targeted delivery of pentagalloyl glucose reverses elastase-induced abdominal aortic aneurysm and restores aorta to the healthy state in mice. PLoS ONE 2020, 15, e0227165. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Parasaram, V.; Dhital, S.; Nosoudi, N.; Hasanain, S.; Lane, B.A.; Lessner, S.M.; Eberth, J.F.; Vyavahare, N.R. Systemic delivery of targeted nanotherapeutic reverses angiotensin II-induced abdominal aortic aneurysms in mice. Sci. Rep. 2021, 11, 8584. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular Calcification in Animal Models of CKD: A Review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Babic, M.; Tölle, M.; van der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney Dis. 2004, 44, 1024–1030. [Google Scholar] [CrossRef]
- Kramer, H.; Toto, R.; Peshock, R.; Cooper, R.; Victor, R. Association between Chronic Kidney Disease and Coronary Artery Calcification: The Dallas Heart Study. J. Am. Soc. Nephrol. 2005, 16, 507–513. [Google Scholar] [CrossRef]
- Neven, E.; Opdebeeck, B.; De Maré, A.; Bashir-Dar, R.; Dams, G.; Marynissen, R.; Behets, G.J.; Verhulst, A.; Riser, B.L.; D’Haese, P.C. Can Intestinal Phosphate Binding or Inhibition of Hydroxyapatite Growth in the Vascular Wall Halt the Progression of Established Aortic Calcification in Chronic Kidney Disease? Calcif. Tissue Int. 2016, 99, 525–534. [Google Scholar] [CrossRef]
- Bowley, G.; Kugler, E.; Wilkinson, R.; Lawrie, A.; van Eeden, F.; Chico, T.J.A.; Evans, P.C.; Noël, E.S.; Serbanovic-Canic, J. Zebrafish as a tractable model of human cardiovascular disease. Br. J. Pharmacol. 2022, 179, 900–917. [Google Scholar] [CrossRef]
- Isogai, S.; Horiguchi, M.; Weinstein, B.M. The Vascular Anatomy of the Developing Zebrafish: An Atlas of Embryonic and Early Larval Development. Dev. Biol. 2001, 230, 278–301. [Google Scholar] [CrossRef] [Green Version]
- Margiotta-Casaluci, L.; Owen, S.F.; Rand-Weaver, M.; Winter, M.J. Testing the Translational Power of the Zebrafish: An Interspecies Analysis of Responses to Cardiovascular Drugs. Front. Pharmacol. 2019, 10, 893. [Google Scholar] [CrossRef] [Green Version]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don’t stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Hoareau, M.; El Kholti, N.; Debret, R.; Lambert, E. Zebrafish as a Model to Study Vascular Elastic Fibers and Associated Pathologies. Int. J. Mol. Sci. 2022, 23, 2102. [Google Scholar] [CrossRef]
- Apschner, A.; Huitema, L.F.A.; Ponsioen, B.; Peterson-Maduro, J.; Schulte-Merker, S. Zebrafish enpp1 mutants exhibit pathological mineralization, mimicking features of generalized arterial calcification of infancy (GACI) and pseudoxanthoma elasticum (PXE). Dis. Models Mech. 2014, 7, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.S.; Küçükosmanoğlu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef] [Green Version]
- Mackay, E.W.; Apschner, A.; Schulte-Merker, S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development 2015, 142, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Jinnouchi, H.; Sato, Y.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Kawakami, R.; Gadhoke, N.V.; Kolodgie, F.D.; Virmani, R.; Finn, A.V. Calcium deposition within coronary atherosclerotic lesion: Implications for plaque stability. Atherosclerosis 2020, 306, 85–95. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van den Branden, A.; Verhulst, A.; D’Haese, P.C.; Opdebeeck, B. New Therapeutics Targeting Arterial Media Calcification: Friend or Foe for Bone Mineralization? Metabolites 2022, 12, 327. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12040327
Van den Branden A, Verhulst A, D’Haese PC, Opdebeeck B. New Therapeutics Targeting Arterial Media Calcification: Friend or Foe for Bone Mineralization? Metabolites. 2022; 12(4):327. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12040327
Chicago/Turabian StyleVan den Branden, Astrid, Anja Verhulst, Patrick C. D’Haese, and Britt Opdebeeck. 2022. "New Therapeutics Targeting Arterial Media Calcification: Friend or Foe for Bone Mineralization?" Metabolites 12, no. 4: 327. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12040327