A Review of Cardiovascular Toxicity of Microcystins


Department of Occupational and Environmental Health, Xiangya School of Public Health, Central South University, Changsha 410078, Hunan, China
*
Author to whom correspondence should be addressed.
Toxins 2019, 11(9), 507; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090507
Submission received: 16 July 2019
/
Revised: 27 August 2019
/
Accepted: 28 August 2019
/
Published: 30 August 2019
Abstract
:The mortality rate of cardiovascular diseases (CVD) in China is on the rise. The increasing burden of CVD in China has become a major public health problem. Cyanobacterial blooms have been recently considered a global environmental concern. Microcystins (MCs) are the secondary products of cyanobacteria metabolism and the most harmful cyanotoxin found in water bodies. Recent studies provide strong evidence of positive associations between MC exposure and cardiotoxicity, representing a threat to human cardiovascular health. This review focuses on the effects of MCs on the cardiovascular system and provides some evidence that CVD could be induced by MCs. We summarized the current knowledge of the cardiovascular toxicity of MCs, with regard to direct cardiovascular toxicity and indirect cardiovascular toxicity. Toxicity of MCs is mainly governed by the increasing level of reactive oxygen species (ROS), oxidative stress in mitochondria and endoplasmic reticulum, the inhibition activities of serine/threonine protein phosphatase 1 (PP1) and 2A (PP2A) and the destruction of cytoskeletons, which finally induce the occurrence of CVD. To protect human health from the threat of MCs, this paper also puts forward some directions for further research.
Key Contribution: For the first time, the manuscript highlights the role of microcystins and cardiovascular outcome. It is a comprehensive review and will be a good read for the cardiovascular researchers.
1. Introduction
The cardiovascular system is considered a dynamic system. As the first organ in embryonic development, the heart provides nutrients and oxygen to all of the various organs and tissues. The normal functions of the cardiovascular system are mainly affected by genetic factors, environmental factors and the interaction between the two factors, which is considered a principal cause of cardiovascular disease (CVD) [1]. It is estimated that about 290 million patients suffer from CVD [2]. Additionally, the recent epidemiological survey indicated that the prevalence and mortality of CVD in China is still on the rise, and the mortality rate of CVD is the highest, in comparison to tumors and other diseases, accounting for more than 40% of the total number of deaths resulting from diseases in China [2]. The burden of CVD is increasing, which has become a major public health problem. Thus, it is important to take measures towards preventing and curing of CVD.
The increasing eutrophication and frequent cyanobacterial blooms in water bodies have become a severe public health concern globally. Cyanobacterial blooms are usually characterized by the mass formation of cyanobacteria, green algae and diatom, leading to the death of aquatic organisms due to hypoxia. In addition, some algae (blue-green algae) including Microcystis, Anabaena, Anabaenopsis, Oscillatoria and Nostoc [3,4,5,6] can produce various potent toxins, such as cyclic peptides, alkaloids and lipopolysaccharides, in the process of metabolism [7]. Subsequently, negative human and animal health effects may be induced when exposed to these toxins [8,9]. Among the cyanotoxins produced during cyanobacterial blooms, monocyclic peptide microcystins (MCs) are the most abundant, widely distributed, extremely toxic and difficult to remove when dissolved in water bodies [10,11].
The basic chemical structure of MCs is cyclo-(D-Ala 1-X 2-D-MeAsp 3-Z 4-Adda 5-D-Glu 6-Mdha 7) (as shown in Figure 1A). In addition, (2S, 3S, 8S, 9S)-3-Amino-9-methoxy-2,6,8-trimethyl-10-phenyldec-4,6-dienoic acid (Adda) is critical for the toxicity of MCs, since this region is found only in cyanobacterial peptides (MCs and nodularins) and is vital for the interactions with protein phosphatases. X and Z in positions 2 and 4 are variable L amino acids. D-MeAsp 3 is D-erythro-β-methylaspartic acid and Mdha is N-methyldehydroalanine [12]. The alterations in amino acids have resulted in the identification of over 200 MC isomers [13]. The main differences lie in the two L-amino acids at positions 2 and 4 as well as the methylation or demethylation of D-Me Asp and M-Dha. The most common congeners are MC-LR (Microcystin-LR), MC-RR (Microcystin-RR) and MC-YR (Microcystin-YR), with different combinations of Leucine (L), arginine (R) or tyrosine (Y) (as shown in Figure 1B). MC-LR is the most toxic isomer with a contribution of 46–99.8 % of the total MCs in natural waters [14,15,16]. Although MC-RR and MC-YR are also widely distributed, their toxicities are second to MC-LR [17,18].
Studies have shown that human beings are exposed to MCs mainly through drinking water, body contact, long-term use of seafood and algal dietary supplements, where drinking water is the principal route [8]. To minimize the health hazard caused by MCs, the World Health Organization (WHO) specifies that the maximum allowable content of MCs in drinking water should not exceed 1 μg/L [20]. Research indicated that MCs negatively affect various human organs when exposed to it. Several organs or tissues including liver [21,22,23,24,25,26], kidney [25,26,27], nervous system [28,29,30], gastrointestinal tract [31,32,33], reproductive system [34,35] and cardiovascular [36] have been reported to be target organs for MC toxicity. This review will focus on the cardiovascular toxicity of MCs and provide some evidence of cardiovascular toxicity caused by MCs. To protect human health from the threat of MCs, this paper also puts forward some directions for further research.
2. Cardiovascular Toxicity of MCs
To date, a large number of studies have shown that MCs can induce cardiovascular toxicity in vivo and vitro directly [36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. Table 1 and Table 2 represent summaries of cardiovascular toxicity in in vivo and vitro studies, respectively. That is, MCs have the potential to cause CVD directly. Numerous investigators have also demonstrated that MCs are capable of causing CVD by inducing pathological changes in structure and/or function of other target organs or tissues [21,22,23,24,25,26,27,31,32,33,54,55,56,57,58,59,60,61,62], namely indirect cardiovascular toxicity of MCs. Figure 2 illustrates the possible mechanisms of indirect cardiovascular toxicity induced by MCs.
Abbreviations in Table 1 and Table 2: SDH: succinate dehydrogenase; MMP: mitochondrial membrane potential; LDH: lactic dehydrogenase; AST: aspartate aminotransferase; CK: creatine kinase; GSH: glutathione; GPx: glutathione peroxidase; ALP: alkaline phosphatase; ALT: alanine aminotransferase; RBC: red blood cell counts; MCH: mean corpuscular hemoglobin; MCV: mean corpusular volume; MCHC: mean corpusular hemoglobin concerntration; HGB: hemoglobin; HCT: red blood cell specific volume; WBC: white blood cell counts; Mon: monocyte; Gran: granulocyte; PCNA: proliferating cell nuclear antigen; ROS: reactive oxygen species; SOD: superoxide dismutase; MDA: malondialdehyd; NRF 2: erythroid-like 2; HO 1: heme oxygenase 1; CAT: catalase; GST: glutathione s-transferase; GLT: glutamate transporter; AchE: acetyl cholinesterase.
2.1. Direct Cardiovascular Toxicity
This implies that MCs can affect the cardiovascular system, including all tissues, cells, blood and vascular in the heart, directly, and result in abnormal structure and/or functioning of the cardiovascular system.
2.1.1. Transportation of MCs
Researchers have demonstrated that MCs can be transported into the various cells through organic anion transporting polypeptides (OATP) after exposure [63]. Almost every organ is capable of expressing the OATP family genes [8,64], though some OATP genes are expressed preferentially in specific tissues, or even selectively expressed [65]. Heart tissue is no exception [66]. Several OATP family genes including OATP4A1, OATP2A1, OATP2B1 and OATP3A1 have been reported to be expressed in heart tissue [67,68]. This signifies that MCs can be transported to and accumulated in cardiac tissue cells, and finally jeopardizing them, though the mechanisms of MCs entering cardiomyocytes await further exploration [69]. Moreover, MCs have been confirmed to enter into the cell depending on the degree of blood perfusion and types and expression levels of OATP carriers [70].
2.1.2. Cytoskeleton Disruption and Mitochondrial Dysfunction
The cardiomyocytes, fibroblasts, telocytes, mast cells, endothelial cells, white blood cells and other immunologically cells (including smooth muscle cells, adipocytes and pericytes) are documented as cells in the cardiac tissue [71]. Among the cells, cardiomyocytes are reported to be rich in mitochondria, accounting for about 40% of the volume of myocardial cells [71]. Kowaltowski et al. demonstrated that the large number of unsaturated fatty acids on the mitochondrial membrane made cardiomyocytes vulnerable to free radicals and prone to the occurrence of oxidative stress [72]. Oxidative stress is related to the pathophysiology of many cardiomyopathies, such as anthracycline mediated cardiomyopathy [73] and alcoholic cardiomyopathy [74]. Goffart et al. declared that mitochondrial defects contributed to cardiomyopathy and heart failure (HF) [75]. An increasing number of studies have also confirmed that MCs can cause oxidative stress imbalance in mitochondria, further resulting in infiltration of neutrophils in tissues, raising secretion of protease and producing substantial oxidative intermediates, thus conducing to cell aging and even death [76,77,78,79,80].
In 2001, for the first time, Zhang et al. [37] proved that MC-LR could induce cardiotoxicity. Short-term toxic effects of MC-LR on SD (Sprague-Dawley) rats by intraperitoneal (I.P.) injection with different doses of MC-LR was investigated, and the results indicated that MC-LR could damage the physical structure of cardiomyocytes and alter biochemical parameters, including lactic dehydrogenase, aspartate aminotransferase and creatine kinase (CK) [37]. Subsequently, a follow up study was conducted and MC-LR was confirmed to have an involvement in pericardial edema and tubular heart formation in loach, Misguruns mizolepis Gunthe embryos [38]. In a chronic study [39], Wistar rats were injected with 10 μg/kg MC-LR by I.P. every two days for eight months and MC-LR treated animals exhibited no remarkable change in appearance and morphology of the heart. Although the results of TUNEL (terminal deoxynucleotide transferase-mediated deoxy-UTP nick end labelling) assay demonstrated no alteration in apoptosis, the cytoskeletons of cardiomyocytes were destructed, which were characterized by a loss of cell crossstriations, lower myofibril volume fraction, enlargement of cardiomyocytes volume, decrease of myofibrillar volume and even fibrosis, and infiltration of mononuclear in the interstitial tissue [39]. In 2010, a similar chronic study was carried out using MC-YR by Suput et al. [40]. Results showed that the volume and density of myocardium decreased with fiberous proliferation, and a few of them were infiltrated by lymphocytes. In addition, larger cardiomyocytes and abnormal nuclear structure were found, but the TUNEL assay results revealed no increase in apoptosis. Taken together, these results suggested that long-term exposure to relatively low doses of MC-LR and MC-YR can induce myocardial atrophy and fibrosis.
Acute exposure to MCs has also been demonstrated to induce cytoskeleton disruption and mitochondrial dysfunction. In an attempt to investigate the toxicity of MC-LR to the heart by I.P. injection into rats with concentrations 0.16 LD50 (14 g/kg) and 1 LD50 (87 g/kg), Qiu et al. [41] reported myocardial infarction in almost all dead rats, and the surviving rats displayed a decrease in heart rate and blood pressure, which tend out to be associated with myocardial mitochondrial dysfunction. Further microscopic examination of pathological ultrastructure showed loss of adhesion between cardiomyocytes and swelling or rupture of mitochondria, and biochemical index test exhibited raising levels of CK and troponin I, which suggested an existence of cardiomyocytes damage. In addition, the level of lipid peroxide was notably increased, prompting the occurrence of severe mitochondrial oxidative stress. Additionally, the respiratory chain enzyme complexes I and III were found to be inhibited, indicating that the mitochondrial electron transfer chain was blocked. Zhao et al. [36] injected MC extracts (mainly containing MC-LR and MC-RR) into the abdominal cavity of rabbits at the dose of 12.5 and 50 μg MC-LR eq/kg bw (body weight) and detected the ultrastructure and enzyme activity in mitochondria after hours of injection. Morphological changes of cardiac mitochondria, increased concentrations of lipid peroxide and activity of succinate dehydrogenase were observed. Nicotinamide adenine dinucleotide (NADH) dehydrogenase was also found to be inhibited, which further affected the mitochondrial electron transfer chain. Moreover, MCs could alter the activities of Ca2+-Mg2+-ATPase in mitochondria, thus destroying the ion homeostasis, which may conduce to the loss of mitochondrial membrane potential (MMP), and finally succeed in damnifying mitochondria of myocardial cells.
Wang et al. in a recent publication have provided a new interpretation of the possible role of MCs in the toxicological mechanisms of vascular dysplasia in vivo and in vitro [42]. Zebrafish juvenile and human umbilical vein endothelial cells (HUVECs) were co-cultured with MC-LR at the dose of 0.1 µM and 1 µM respectively. In vivo, MC-LR resulted in angiodysplasia, damaged vascular structures, reduced lumen size and blood flow as well as vascular dysfunction, while in vitro, apoptosis, activity of caspase3/9, mitochondrial ROS and p53 were increased, whereas MMP and proliferating cell nuclear antigen were inhibited. To explore the effect of MC-LR on cardiopulmonary system, Martins et al. [43] injected 100 μg MC-LR/kg into trahira, Hoplias malabaricus and reported that MC-LR induced cytotoxicity by enhancing oxidative stress and activity of related enzymes, and consequently affected cardiopulmonary function. This was similar to what Qiu et al. demonstrated [41]. In addition, MC-LR has recently been confirmed to inhibit the heart rate of Japanese Medaka (Oryzias Latipes) [44]. Currently, Xu et al. [49] provided an explanation of the possible role of antioxidant enzymes in the toxicological mechanisms of MCs at the transcription level by treating H9C2 (a kind of rat cardiomyocyte) cell lines with 10 μM MC-LR. The expression levels of cardiac rhythm and antioxidant genes were detected and the results suggested that the expression levels of rhythmic genes (baml1, cry1, cry2, per1, per2) were inhibited, while the antioxidant genes (catalase, ho-1, sod1, sod2) were upregulated. These results indicated that alteration in the rhythm of cardiomyocytes is one of the possible cardiac toxilogical mechanisms of MCs.
The above evidence suggests that MCs can directly lead to cardiac malformation in larva, and induce myocardial atrophy and fibrosis in adults, although the mechanisms of the effect need to be further explored. MCs may also induce cardiovascular toxicity by transforming the morphology of cardiomyocytes, cell proliferation and apoptosis, cytoskeleton and cell rhythm, as well as the ultrastructure, oxidative stress, membrane potential and enzyme activity of respiratory chain in mitochondria of cardiomyocytes.
2.1.3. Endoplasmic Reticulum Dysfunction
MCs are also known to give rise to CVD by damaging the endoplasmic reticulum (ER). ER is a tunneling system composed of membranes in eukaryotic cells, and it is an important organelle for protein synthesis, folding and secretion [81,82]. The stability of the ER environment is an essential precondition for realizing ER function. When the internal or external microenvironment of ER changes, the imbalance function of homeostasis may be caused, resulting in the occurrence of endoplasmic reticulum stress response (ERS) [83,84]. Ischemia-reperfusion injury, homocysteine and other chemicals’ treatment, protein synthesis abnormality, protein folding capacity dysregulation, ER calcium metabolism disorder, physicochemical or genetic factors, such as the disturbance of lecithin synthesis, are capable of arousing ERS [85,86]. Moderate stress was able to protect cells through unfolded protein response, while prolonged or excessive stress could trigger ER CHOP, JNK and Caspase-1/2, Ca2+ pathways to induce apoptosis [87]. This suggests that cardiomyocytes damage can be produced by interfering with ER stress-related pathways. Previous data indicated that ERS evoked from lipid overload, changes in redox state, free radicals and other physical and/or chemical factors, could cause apoptosis of endothelial cells and monocytes entering the vascular endothelium to engulf lipid and lipid foam cells formation, thus giving rise to CVD [88]. In summary, the occurrence and development of CVD such as atherosclerosis [89,90], diabetic heart disease [91], hypertension [92,93], myocardial hypertrophy and HF [94,95] are closely related to ERS.
In recent years, a considerable number of studies have demonstrated that MC-LR can induce ERS. Qi et al. [45] found deformed morphology, stunted growth, suppressed heart rate and increased apoptosis when zebrafish juveniles were treated with 4.0 mM MC-LR for 96 h. Further studies on the mechanism showed that the above phenotypes could be partially rescued by tauroursodeoxycholic acid (TUDCA, 20 mM, an inhibitor of ERS), which suggested that the developmental toxicity of MC-LR was possibly produced by activating ERS. That is, MC-LR could induce developmental toxicity and apoptosis by increasing ER oxidative stress. In addition, Cai et al. [96,97] demonstrated that the raise of ER stress and apoptosis had an involvement in neurotoxicity caused by MC-LR in rats. A study conducted by Zhao et al. [98] provided an explanation of the possible role of oxidative stress and ER stress in the toxicological mechanisms of MCs at the proteomic level, and the results revealed that MC-LR remarkably altered the abundance of 49 proteins that were involved in oxidative phosphorylation, cytoskeleton, metabolism, protein folding and degradation. Although there is no direct evidence that MC-LR can induce ERS in the cardiovascular system, it is reasonable to infer that MC-LR is capable of affecting cardiovascular function by activating ERS.
2.1.4. Inhibition of PP1 and PP2A
Previous research exhibited that the main mechanism of MCs producing their toxic effects is to inhibit serine/threonine protein phosphatase 1 (PP1) and 2A (PP2A) by interacting with the subunits of serine/threonine protein phosphatase (PP), which result in the disruption of the dynamic equilibrium of protein phosphorylation as well as expression and activation of their downstream proteins, and further leading to the cytoskeletal reorganization [8,99,100,101,102,103,104]. PP regulates a series of processes in mammalian cells, including cell proliferation, division, signal transduction and gene expression [105]. In the cardiovascular system, reversible protein phosphorylation is central to a variety of cardiac processes such as excitation–contraction coupling, Ca2+ handling, cell metabolism, myofilament regulation and intercellular communication [100]. PP1 and PP2A have been reported to regulate the cardiac function by dephosphorylating a disparate collection of target proteins, Cav1.2 and ATP sensitive Na+/K+ channels [101,102,106,107,108,109]. Overexpression of PP2A-A subunit or its active substitute PP2B was found to induce myocardial hypertrophy or HF [110,111,112]. Similarly, Meyer-Roxlau et al. [103] in a recent investigation reported that the increasing activity of PP1 contributed to cardiac hypertrophy, HF and atrial fibrillation. Furthermore, PP might as well directly dephosphorylate some signal molecules or transcription factors to give rise to the occurrence of CVD [113,114]. Since a considerable number of data have indicated that MCs inhibited the activities of PP1 and PP2A [8,19,22,29], it is profitable to infer that the inhibition activities of PP1 and PP2A is one of the possible cardiac toxicological mechanisms of MCs.
2.1.5. Hemodynamic Alterations and Vascular Lesions
The heart is known to be rich in vessels and bloods, and vessels are the carrier of blood flow. The heart supplies blood to the body, providing power for the body’s normal metabolism [115,116]. Blood flow alterations and vascular lesions can jeopardize the function of the heart. LeClaire [46] noted a continuous decrease of heart rate, cardiac output (CO), stroke, oxygen consumption, carbon dioxide production, metabolic rate, accompanied progressive hypothermia and disrupted equilibrium of acid–base when male Fischer 344 rats were administered with I.P. injection of MC-LR at a dose of 100 μg/kg bw. Huang et al. [47] investigated the response indices to toxic MC-LR in the blood of mice with I.P. injection at different MC-LR concentrations and indicated that the phagocytic index, ROS, hematology of the majority of blood cells and volume of erythrocytes were influenced by the toxin. In addition, the alterations of some cytokines and ROS of leukocytes were observed [47]. Another in vivo study also showed a significant difference in red blood cell parameters (red blood cell counts, haematocrit values, mean corpuscular hemoglobin, mean corpusular volume and mean corpusular hemoglobin concerntration) when rats were fed with fish meat with and without MCs for 28 days [48]. In addition, in vitro investigation indicated that enzyme concentrations in blood including glutathione, superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) and glutathione s-transferase were altered. Increased hemolysis and pathological alterations in agglomerated and jagged erythrocytes were observed [53]. All these aforementioned results demonstrated that MC-LR possesses blood toxicity.
Vascular endothelial cells are between blood and vascular wall tissue, which are composed of a layer of monocytes. Endothelial cells are the key regulators of vascular dynamic equilibrium and have an involvement in cell mitosis, vascular regeneration, vascular osmotic pressure, inflammatory response and platelet activity [117,118]. Therefore, any change in the structure and/or function of vascular endothelial cells may cause a destruction of the vascular system [119]. HUVECs were treated with MC-LR at a dose of 40 μM for 24 hours and the proliferation, migration ability and capillary-like structure formation of vascular endothelial cells were reported to be inhibited [50,51,52], whereas the apoptosis of endothelial cells, production of ROS, oxidative stress, expression levels of inflammatory factors and endothelial cell adhesion factors were found to be increased [50,51,52]. In another in vivo study, dysplastic, dysfunctional, destroyed structure, narrowed size of vascular and slower blood flow were found when zebrafish juveniles were co-cultured with MC-LR at a dose of 4.0 mM for 96 h [45]. The evidence thus suggests that MC-LR can induce vascular toxic effects.
2.2. Indirect Cardiovascular Toxicity
The functions of mammalian organs usually intertwine. These organs are related to each other and affect and restrict each other. This implies that a dysfunction in one organ may cause a dysfunction in another organ or the abnormality of one organ may affect the normal function of another organ [54,55,56,57,58,59,60,61,62]. Similarly, MCs are capable of causing further CVD by inducing pathological changes in the structure and/or function of other target organs or tissues [21,22,23,24,25,26,27,31,32,33,54,55,56,57,58,59,60,61,62]—that is, the indirect cardiovascular toxicity of MCs. The following are studies demonstrating how abnormalities in other organs give rise to CVD.
2.2.1. Liver Diseases Induced by MCs and CVD
The liver is one of the most vital target organs of MCs. MCs are transported into hepatocytes through OATP1B1 and OATP1B3, and exert hepatotoxicity [120]. A study carried out by Falconer et al. [121] showed that, under the induction of MC-LR, the cytoskeleton damage of isolated hepatocytes resulted in the loss of cell morphology, cell adhesion and cell death. The potential mechanism might be the inhibition of phosphatase, invoking hyperphosphorylation in a large number of hepatocytes, including keratin, which is responsible for microfilament orientation and intermediate filament integrity. As mentioned earlier, MC-LR can irreversibly inhibit the activities of PP1 and PP2A in hepatocytes, and may account for the disintegration of hepatocytes, apoptosis, necrosis, nonalcoholic steatohepatitis disease (NASH), intrahepatic hemorrhage, liver cancer and even animal death [122,123]. MC-LR may also induce hepatotoxicity by inhibiting the survival rate of hepatocytes, increasing hepatocyte apoptosis and ROS levels [124,125] as well as the activities of SOD, CAT, GPx and glutathione reductase in hepatocytes [126,127]. Several studies have shown that liver diseases are closely related to CVD. In a recent population survey, Mellinger et al. [128] reported that NASH may contribute to CVD including hypertension and atherosclerosis. Clinical data exhibited that NASH patients had higher carotid intima media thickness [129,130] and often experienced altered cardiac structure [131,132,133]. In addition, Boddi et al. [134] found that patients with ST-elevation myocardial infarction more often than not experienced NASH. The data suggest that MC-LR may induce NASH which has the potential to conduce CVD.
Krag et al. investigated 24 patients with liver cirrhosis and reported an increase in cardiac volume load and end-diastolic volume, as well as a decrease in left ventricular wall motion, CO and ejection fraction. Interestingly, myocardial perfusion was preserved [54]. Glenn et al. also exhibited a significant decline in diastolic reflux velocity, prolonged diastolic time and increased passive tension by assessing the relationship between liver diseases and CVD in cirrhotic animals [135]. Thus, liver disease is able to disrupt the cardiovascular system to cause diastolic dysfunction leading to CVD development. In a serum survey, Henriksen et al. [55] examined 51 patients with liver cirrhosis and revealed higher pro-BNP (pro-brain natriuretic peptide) and BNP (brain natriuretic peptide) in serum than those in normal subjects. Pro-BNP and BNP were generally indicators of abnormal QT-interval, heart rate and plasma volume in cardiac function [55]. In a recent study, Schimmel et al. [136] found that BNP levels in serum were related to cardiac function and severity of HF in patients with chronic heart failure (CHF). BNP levels were increased when cardiac systolic or diastolic dysfunction occurred, respectively, and, when both existed at the same time, the increasing levels were more significant [136]. Dong et al. [137] have also demonstrated that BNP levels in serum of cirrhosis were associated with ventricular wall thickness, diastolic dysfunction, stress-induced systolic dysfunction, hyperdynamic circulation and cardiac structural changes. Additionally, inflammation of liver, oxidative stress level of hepatocytes and apoptosis and proliferation of hepatocytes are able to increase the risk of CVD [138,139]. The evidence thus suggests that liver diseases caused by MCs can alter cardiac contractility, cardiac diastolic function and electrocardiograms through a variety of mechanisms, including autonomic nerve regulation, inflammation and changes in membrane channels, in order to give rise to CVD.
2.2.2. Intestinal Diseases Induced by MCs and CVD
The gastrointestinal tract is another target organ of MCs. Oral exposure to MCs is the principal route of exposure [10]. After ingestion, MCs are absorbed into the gastrointestinal tract, transported into gastrointestinal epithelial cells mainly through OATP3A1 and OATP4A1 [31], and finally exert their multi-organ and multi-tissue toxicity through blood circulation. Studies revealed that partially unabsorbed MCs accumulated in the gastrointestinal tract can exert gastrointestinal toxic effects [32,33]. The earliest investigation on the intestinal toxicity of MC-LR was done by Falconer et al. [140] in 1992, by treating intestinal cells isolated from chickens with MCs. The intestinal cells exhibited time-and dose-dependent deformation or even death, and one or more blisters grew on the surface of deformed cells after the induction of MCs. The study also pointed out that the production of gastroenteritis related to ingestion of MCs may reflect the injury of intestinal epithelial cells caused by MCs. Botha et al. also in an in vitro study displayed that MC-LR could decrease the viability and induce time-dependent apoptosis of CaCo2 cells (an intestinal cell line) after the cell line was exposed to 50 μM/L MC-LR [141]. Furthermore, an acute in vivo study indicated that MC-LR could induce time-dependent apoptosis of duodenal, jejunum and ileum cells [78]. In view of this, MCs may have the ability to induce time-and dose-dependent toxic effects on the gastrointestinal tract.
Results of immunohistochemistry in vivo studies exhibited that MC-LR was mainly accumulated in intestinal microvilli [78], mucous layer, villi epithelial, lamina propria cells, and mucus secreted by the goblet cells of the small intestine [32], and in the cytoplasm and around the nucleus [142]. A comparative research explored by Gaudin et al. [143] adopted the method of feeding and I.P. injection of MC-LR for mice simultaneously. Comet assay suggested that both routes of exposure could remarkably increase the damage of intestinal DNA in mice, whereas the toxicity of I.P. injection of MC-LR was more serious. In summary, current explorations have shown that MCs induce gastrointestinal toxicity mainly by destroying the physical structure [143], the immune system [144], the balance of water and electrolytes in cells [143] and altering the activity of digestive enzymes in the chorion of the intestine [145], or inducing oxidative stress and apoptosis in intestinal cells [146,147,148] or even altering the intestinal microbes [149,150]. Meanwhile, there is considerable evidence that patients with inflammatory intestinal diseases have a higher risk of CVD [56,57]. In the early 1990s, Levine et al. [151] reported that HF belonged to a kind of chronic inflammation, and the expression levels of pro-inflammatory factors in serum were closely concerned with the morbidity rate of HF. Anker et al. [152] also indicated that a large number of inflammatory factors first originated from the intestinal tract. This suggested that the destruction of intestinal physical structure and immune system might transform the permeability of intestinal epithelial cells, and be conducive to uncontrolled entry and exit of substances into and out of cells, and finally flow into the various organs through blood circulation, leading to CVD. Data in recent years showed that animals displaying intestinal microbial alterations, bacterial translocation and the presence of bacteria in circulation after the destruction of intestinal barrier function brought about CVD such as vasculitis, hypertension, atherosclerosis and CHF [153,154,155].
2.2.3. Kidney Diseases Induced by MCs and CVD
Although MCs are mainly accumulated in the liver and discharged through the bile duct, a small part of these toxins (about 9%) is filtered in the kidney and discharged through urine [156], which makes the kidney a potential target for MC toxicity. MCs can enter the kidney through OATP1 [157]. Menezes et al. [158] and Jia et al. [159] proved that MC-LR was capable of accumulating in the kidney and manifested its nephrotoxic effects. In a recent in vivo study, Wang et al. [160] confirmed the nephrotoxic toxicity induced by MC-LR. Zebrafish were treated with different doses of MC-LR and the pathological alterations in kidney tissue showed the existence of eosinophilic casts in renal tubules, abnormal renal tubules, decreased space intertubular and blood infiltration in renal cells. RNA-Seq analysis indicated disrupted renal gene expressions that had some involvement in various pathways, such as oxidative phosphorylation, cell cycle and protein processing in ER, concerned with apoptosis. TUNEL assay found the presence of renal cell apoptosis. Additionally, negative changes in the ROS level, apoptotic-related gene, protein expressions and enzyme activities revealed that MC-LR could induce production of ROS, subsequently triggering apoptosis via p53-bcl-2 and caspase-dependent pathway in the kidney. The data signifies that apoptosis may be a primary case of MC-LR-induced nephrotoxicity. Similarly, Nicole et al. in a recent review of the toxicology mechanism of MCs claimed that chronic exposure to low doses of MCs may pose a great risk to nephrotoxicity in mammals. MC-LR induced renal dysfunction, vascular and glomerular lesions and alterations in kidney tissues mainly by disrupting mitochondria and increasing ROS levels [161].
An in vitro exploration conducted by Dias et al. [162] demonstrated that MC-LR could induce the proliferation of renal cells through p38, JNK and Erk1/2 signaling pathways, leading to the emergence of renal tumors, after the monkey kidney cell line Vero-E6 were treated with different concentrations of MC-LR. Similarly, the human embryonic kidney 293 cell line (HEK293) and human kidney adenocarcinoma cell line (ACHN) were treated with different doses of MC-LR for 24 hours, and decreased viability as well as increased apoptosis of both cell lines were observed [27]. This suggests that MC-LR may contribute to chronic kidney disease (CKD) by damaging the renal cells.
Previous studies have shown that CVD is the most serious complication in patients with CKD, and its incidence rate is also increasing year by year. It is also the main cause of death in patients with CKD [58,59]. A cohort survey of patients with CKD indicated that CKD patients had higher blood pressure [60] and higher expression levels of biomarkers of inflammation-related factors, such as C-reactive protein, interleukin-6 and higher levels of endotoxin in serum, which can increase hemodynamic load and result in volume overload [61]. Moreover, due to long-term CKD, an iron metabolism disorder hinders the differentiation of red blood cells, and renal erythropoietin also decreases, leading to severe anemia in CKD patients [62]. A long-term increase in blood pressure, high expression levels of inflammatory factors, high volume load and anemia can inhibit the activation of xanthine oxidase, NADPH oxidase uncoupled nitric oxide synthase, and finally succeed in inducing oxidative stress as well as ROS level, decreasing antioxidant defense ability, nitric oxide and its activity, which give rise to left ventricular hypertrophy, vascular endothelial dysfunction, atherosclerosis and other CVD such as cardiac remodeling / fibrosis and HF [62,134].
3. Conclusions and Outlook
In this current paper, the effects of MCs on cardiovascular system were reviewed. Those studies have shown that cardiovascular toxicity is closely associated with MC exposure at various doses, pathways and in different species. Figure 3 outlines the possible mechanisms of cardiovascular toxicity of MCs. After exposure, MCs are transported into cells via OATP, and accumulated in the main organs and tissues. At this stage, MCs are preferentially concentrated in the liver, gastrointestinal tract, kidneys, cardiovascular and other organs. Having entered the cardiovascular system, MCs can induce cardiovascular toxicity directly by altering the morphology of cardiomyocytes, state of cellular apoptosis and proliferation, cytoskeleton, rhythm, the differential expression/activity of transcription factors, the ultrastructure, MMP and enzyme activity in respiratory chain in mitochondria of cardiomyocytes. MC exposure is also capable of raising the production of ROS and ER oxidative stress, resulting in cytoskeleton destruction, mitochondrial dysfunction and ER dysfunction. In addition, MCs are able to inhibit the activities of PP1 and PP2A, which lead to hyperphosphorylation of regulatory proteins, thus regulating cytoskeleton tissue, cell proliferation, apoptosis and CVD. MC exposure might also be associated with the damages of myocardial intercellular connection and vascular endothelial cellular cytoskeleton, or the reducing of cell vitality and increasing of apoptosis, oxidative stress levels as well as ROS levels in mitochondria and ER of blood cells and vascular endothelial cells. In addition to cardiovascular direct effects, MCs can indirectly induce CVD by destroying the structure and/or function of other organs including the liver, gastrointestinal area and kidneys. The review showed that exposure to MCs has influential toxic effects on the cardiovascular system of animals. Although there is no human data on cardiovascular toxicity of MCs, it is believed that cardiovascular toxicity caused by MCs may pose a great threat to human health, especially in view of the wide distribution and spread of MCs in the environment.
It is of interest that the gap in understanding cardiovascular toxicity of MCs needs to be addressed through further research. 1. Direct data of exposure to MCs and the identification of MCs in human serum are necessary for human epidemiological studies of cardiovascular effects so as to understand the toxicology mechanism. 2. Whether maternal exposure to MCs during pregnancy affects the development of the heart of the offspring needs further exploration. 3. Do other congeners of MCs also have the potential to induce cardiovascular diseases in addition to MC-LR, MC-YR and MC-RR? 4. Other environmental factors including heavy metals, trace elements and organic pollutants could intensify or attenuate the toxicity of MCs. 5. Advanced and accessible technologies are essential to degrade and remove MCs from water. 6. States should be encouraged to establish acceptable maximum concentrations of MCs in drinking water, recreational water and irrigation water, especially in remote areas where rivers, lakes and streams are their main sources of water. Public education on the toxic effects of MCs and their related diseases should be strengthened. 7. Effective drugs that inhibit the binding of MCs to PP should be invented and manufactured. 8. Due to the elusive mechanism of cardiovascular toxicity of MCs, it is necessary to explore its molecular toxicology mechanism to develop targeted drugs. In addition, the toxicity of MCs to cardiac development needs to be further studied in order to prevent the occurrence of congenital heart disease.
Author Contributions
F.Y. and L.C. conceived and designed the article; L.C. and H.F. designed the figures; L.C., I.Y.M. and F.Y. wrote the paper. All authors participated in the preparation of the manuscript.
Funding
This work was supported by the National Natural Science Foundation (81773393, 81502787), the Central South University Innovation Driven Project (20170027010004), Key Research and Development Projects in Hunan Province (2018WK2013), and the National Key Research and Development program of China (2016YFC0900800).
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- Vecoli, C.; Pulignani, S.; Foffa, I.; Andreassi, M.G. Congenital heart disease: The crossroads of genetics, epigenetics and environment. Curr. Genom. 2014, 15, 390–399. [Google Scholar] [CrossRef]
- Hu, S.; Gao, R.; Liu, L.; Zhu, M.; Wang, W.; Wang, Y.; Wu, Z.; Li, H.; Gu, D.; Yang, Y.; et al. Summary of the 2018 Report on Cardiovascular Diseases in China. Chin. Circ. J. 2019, 34, 209. [Google Scholar]
- Merel, S.; Walker, D.; Chicana, R.; Snyder, S.; Baurès, E.; Thomas, O. State of knowledge and concerns on cyanobacterial blooms and cyanotoxins. Environ. Int. 2013, 59, 303–327. [Google Scholar] [CrossRef]
- Puddick, J.; Prinsep, M.R.; Wood, S.A.; Kaufononga, S.A.; Cary, S.C.; Hamilton, D.P. High levels of structural diversity observed in microcystins from Microcystis CAWBG11 and characterization of six new microcystin congeners. Mar. Drugs 2014, 12, 5372–5395. [Google Scholar] [CrossRef]
- Massey, I.Y.; Zhang, X.; Yang, F. Importance of bacterial biodegradation and detoxification processes of microcystins for environmental health. J. Toxicol. Environ. Health B 2018, 21, 357–369. [Google Scholar] [CrossRef]
- Yang, F.; Massey, I.Y.; Guo, J.; Yang, S.; Pu, Y.; Zeng, W.; Tan, H. Microcystin-LR degradation utilizing a novel effective indigenous bacterial community YFMCD1 from Lake Taihu. J. Toxicol. Environ Health A 2018, 81, 184–193. [Google Scholar] [CrossRef]
- Codd, G.A.; Morrison, L.F.; Metcalf, J.S. Cyanobacterial toxins: Risk management for health protection. Toxicol. Appl. Pharmacol. 2005, 203, 264–272. [Google Scholar] [CrossRef]
- Massey, I.Y.; Yang, F.; Ding, Z.; Yang, S.; Guo, J.; Tezi, C.; Al-Osman, M.; Kamegni, R.B.; Zeng, W. Exposure routes and health effects of microcystins on animals and humans: A mini-review. Toxicon 2018, 151, 156–162. [Google Scholar] [CrossRef]
- Yang, F.; Guo, J.; Huang, F.; Massey, I.Y.; Huang, R.; Li, Y.; Wen, C.; Ding, P.; Zeng, W.; Liang, G. Removal of Microcystin-LR by a Novel Native Effective Bacterial Community Designated as YFMCD4 Isolated from Lake Taihu. Toxins 2018, 10, 363. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Wen, C.; Zheng, S.; Yang, Y.; Feng, X.; Chen, J.; Luo, D.; Liu, R.; Yang, F. Regulation of Microcystin-LR-Induced DNA Damage by miR-451a in HL7702 Cells. Toxins 2019, 11, 164. [Google Scholar] [CrossRef]
- de la Cruz, A.A.; Antoniou, M.G.; Hiskia, A.; Pelaez, M.; Song, W.; O’Shea, K.E.; He, X.; Dionysiou, D.D. Can we effectively degrade microcystins?—Implications on human health. Anticancer Agents Med. Chem. 2011, 11, 19–37. [Google Scholar] [CrossRef]
- Hinojosa, M.G.; Gutierrez-Praena, D.; Prieto, A.I.; Guzman-Guillen, R.; Jos, A.; Camean, A.M. Neurotoxicity induced by microcystins and cylindrospermopsin: A review. Sci. Total Environ. 2019, 668, 547–565. [Google Scholar] [CrossRef]
- Spoof, L.; Catherine, A. Appendix 3. In Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis; Meriluto, J., Spoof, L., Codd, G.A., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 526–537. [Google Scholar]
- Ufelmann, H.; Kruger, T.; Luckas, B.; Schrenk, D. Human and rat hepatocyte toxicity and protein phosphatase 1 and 2A inhibitory activity of naturally occurring desmethyl-microcystins and nodularins. Toxicology 2012, 293, 59–67. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fan, H.; Xie, P.; He, J. A review of neurotoxicity of microcystins. Environ. Sci. Pollut. Res. Int. 2016, 23, 7211–7219. [Google Scholar] [CrossRef]
- Hinojosa, M.G.; Prieto, A.I.; Gutierrez-Praena, D.; Moreno, F.J.; Camean, A.M.; Jos, A. Neurotoxic assessment of Microcystin-LR, cylindrospermopsin and their combination on the human neuroblastoma SH-SY5Y cell line. Chemosphere 2019, 224, 751–764. [Google Scholar] [CrossRef]
- Meriluoto, J.A.; Spoof, L.E. Cyanotoxins: Sampling, sample processing and toxin uptake. Adv. Exp. Med. Biol. 2008, 619, 483–499. [Google Scholar]
- Pearson, L.; Mihali, T.; Moffitt, M.; Kellmann, R.; Neilan, B. On the chemistry, toxicology and genetics of the cyanobacterial toxins, microcystin, nodularin, saxitoxin and cylindrospermopsin. Mar. Drugs 2010, 8, 1650–1680. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.; Zhang, X.; Xie, P. A review of reproductive toxicity of microcystins. J. Hazard. Mater. 2016, 301, 381–399. [Google Scholar] [CrossRef]
- WHO. Cyanobacterial Toxins: Microcystin-LR. In Guidelines for Drinking Water Quality; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Yang, F.; Wen, C.; Zheng, S.; Yang, S.; Chen, J.; Feng, X. Involvement of MAPK/ERK1/2 pathway in microcystin-induced microfilament reorganization in HL7702 hepatocytes. J. Toxicol. Environ. Health Part A 2018, 81, 1135–1141. [Google Scholar] [CrossRef]
- Yang, S.; Chen, L.; Wen, C.; Zhang, X.; Feng, X.; Yang, F. MicroRNA expression profiling involved in MC-LR-induced hepatotoxicity using high-throughput sequencing analysis. J. Toxicol. Environ. Health A 2018, 81, 89–97. [Google Scholar] [CrossRef]
- Hou, J.; Li, L.; Xue, T.; Long, M.; Su, Y.; Wu, N. Hepatic positive and negative antioxidant responses in zebrafish after intraperitoneal administration of toxic microcystin-LR. Chemosphere 2015, 120, 729–736. [Google Scholar] [CrossRef]
- Chen, L.; Hu, Y.; He, J.; Chen, J.; Giesy, J.P.; Xie, P. Responses of the Proteome and Metabolome in Livers of Zebrafish Exposed Chronically to Environmentally Relevant Concentrations of Microcystin-LR. Environ. Sci. Technol. 2017, 51, 596–607. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Liu, F.; Hou, D.X.; Xu, J.; Zhao, X.; Yang, F.; Feng, X. Prodigiosin Promotes Nrf2 Activation to Inhibit Oxidative Stress Induced by Microcystin-LR in HepG2 Cells. Toxins 2019, 11, 403. [Google Scholar] [CrossRef]
- Wen, C.; Yang, S.; Zheng, S.; Feng, X.; Chen, J.; Yang, F. Analysis of long non-coding RNA profiled following MC-LR-induced hepatotoxicity using high-throughput sequencing. J. Toxicol. Environ. Health A 2018, 81, 1165–1172. [Google Scholar] [CrossRef]
- Piyathilaka, M.A.; Pathmalal, M.M.; Tennekoon, K.H.; De Silva, B.G.; Samarakoon, S.R.; Chanthirika, S. Microcystin-LR-induced cytotoxicity and apoptosis in human embryonic kidney and human kidney adenocarcinoma cell lines. Microbiology 2015, 161, 819–828. [Google Scholar] [CrossRef]
- Caban-Holt, A.; Mattingly, M.; Cooper, G.; Schmitt, F.A. Neurodegenerative memory disorders: A potential role of environmental toxins. Neurol. Clin. 2005, 23, 485–521. [Google Scholar] [CrossRef]
- Feurstein, D.; Kleinteich, J.; Heussner, A.H.; Stemmer, K.; Dietrich, D.R. Investigation of microcystin congener-dependent uptake into primary murine neurons. Environ. Health Perspect. 2010, 118, 1370–1375. [Google Scholar] [CrossRef]
- Ding, J.; Wang, J.; Xiang, Z.; Diao, W.; Su, M.; Shi, W.; Wan, T.; Han, X. The organic anion transporting polypeptide 1a5 is a pivotal transporter for the uptake of microcystin-LR by gonadotropin-releasing hormone neurons. Aquat. Toxicol. 2017, 182, 1–10. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, R.; Ye, J.; Wu, X.; Zhang, Y.; Wu, C. Monitoring and research of microcystins and environmental factors in a typical artificial freshwater aquaculture pond. Environ. Sci. Pollut. Res. Int. 2018, 25, 5921–5933. [Google Scholar] [CrossRef]
- Ito, E.; Kondo, F.; Harada, K. First report on the distribution of orally administered microcystin-LR in mouse tissue using an immunostaining method. Toxicon 2000, 38, 37–48. [Google Scholar] [CrossRef]
- Cao, L.; Huang, F.; Massey, I.Y.; Wen, C.; Zheng, S.; Xu, S.; Yang, F. Effects of Microcystin-LR on the Microstructure and Inflammation-Related Factors of Jejunum in Mice. Toxins 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, S.; Liu, C.; Wu, J.; Wang, Y.; Yuan, L.; Du, X.; Wang, R.; Marwa, P.W.; Zhuang, D. Resveratrol Ameliorates Microcystin-LR-Induced Testis Germ Cell Apoptosis in Rats via SIRT1 Signaling Pathway Activation. Toxins 2018, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhuang, H.; Yang, H.; Xue, W.; Wang, L.; Wei, W. Microcystin-LR disturbs testicular development of giant freshwater prawn Macrobrachium rosenbergii. Chemosphere 2019, 222, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, P.; Tang, R.; Zhang, X.; Li, L.; Li, D. In vivo studies on the toxic effects of microcystins on mitochondrial electron transport chain and ion regulation in liver and heart of rabbit. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2008, 148, 204–210. [Google Scholar] [CrossRef]
- Zhang, Z.; Kang, S.; Chen, C.; Wei, G.; Yu, S. The acute toxic effects of microcystin LR in SD rats. Zhonghua Yu Fang Yi Xue Za Zhi 2002, 36, 295–297. [Google Scholar]
- Liu, Y.; Song, L.; Li, X.; Liu, T. The toxic effects of microcystin-LR on embryo-larval and juvenile development of loach, Misguruns mizolepis Gunthe. Toxicon 2002, 40, 395–399. [Google Scholar] [CrossRef]
- Milutinovic, A.; Zorc-Pleskovic, R.; Petrovic, D.; Zorc, M.; Suput, D. Microcystin-LR induces alterations in heart muscle. Folia Biol. (Praha) 2006, 52, 116–118. [Google Scholar]
- Suput, D.; Zorc-Pleskovic, R.; Petrovic, D.; Milutinovic, A. Cardiotoxic injury caused by chronic administration of microcystin-YR. Folia Biol. (Praha) 2010, 56, 14–18. [Google Scholar]
- Qiu, T.; Xie, P.; Liu, Y.; Li, G.; Xiong, Q.; Hao, L.; Li, H. The profound effects of microcystin on cardiac antioxidant enzymes, mitochondrial function and cardiac toxicity in rat. Toxicology 2009, 257, 86–94. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Guo, J.; Lin, S.; Wang, Y.; Yin, T.; Gregersen, H.; Hu, T.; Wang, G. Microcystin-LR induces angiodysplasia and vascular dysfunction through promoting cell apoptosis by the mitochondrial signaling pathway. Chemosphere 2019, 218, 438–448. [Google Scholar] [CrossRef]
- Martins, N.D.; Yunes, J.S.; McKenzie, D.J.; Rantin, F.T.; Kalinin, A.L.; Monteiro, D.A. Microcystin-LR exposure causes cardiorespiratory impairments and tissue oxidative damage in trahira, Hoplias malabaricus. Ecotoxicol. Environ. Saf. 2019, 173, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.R.; Frenkel, A.; Harke, M.J.; Jankowiak, J.G.; Gobler, C.J.; McElroy, A.E. Effects of Microcystis on development of early life stage Japanese medaka (Oryzias latipes): Comparative toxicity of natural blooms, cultured Microcystis and microcystin-LR. Aquat. Toxicol. 2018, 194, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Dang, Y.; Xu, Q.; Yu, L.; Liu, C.; Yuan, Y.; Wang, J. Microcystin-LR induced developmental toxicity and apoptosis in zebrafish (Danio rerio) larvae by activation of ER stress response. Chemosphere 2016, 157, 166–173. [Google Scholar] [CrossRef] [PubMed]
- LeClaire, R.D.; Parker, G.W.; Franz, D.R. Hemodynamic and calorimetric changes induced by microcystin-LR in the rat. J. Appl. Toxicol. 1995, 15, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, Y.; Xiao, W.; Ye, X.; Zhong, Q.; Gu, K. Comparison of response indices to toxic microcystin-LR in blood of mice. Chemosphere 2013, 92, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Palikova, M.; Ondrackova, P.; Mares, J.; Adamovsky, O.; Pikula, J.; Kohoutek, J.; Navratil, S.; Blaha, L.; Kopp, R. In vivo effects of microcystins and complex cyanobacterial biomass on rats (Rattus norvegicus var. alba): Changes in immunological and haematological parameters. Toxicon 2013, 73, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, X.; Jiang, S.; Men, C.; Xu, D.; Guo, Y.; Wu, J. Microcystin-LR regulates circadian clock and antioxidant gene expression in cultured rat cardiomyocytes. Cell. Mol. Biol. Lett. 2018, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, J.; Zhang, M. Microcystins Induces Vascular Inflammation in Human Umbilical Vein Endothelial Cells via Activation of NF-kappaB. Mediat. Inflamm. 2015, 2015, 942159. [Google Scholar] [CrossRef]
- Shi, J.; Deng, H.; Pan, H.; Xu, Y.; Zhang, M. Epigallocatechin-3-gallate attenuates microcystin-LR induced oxidative stress and inflammation in human umbilical vein endothelial cells. Chemosphere 2017, 168, 25–31. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, M.; Zhang, L.; Deng, H. Epigallocatechin-3-gallate attenuates microcystin-LR-induced apoptosis in human umbilical vein endothelial cells through activation of the NRF2/HO-1 pathway. Environ. Pollut. 2018, 239, 466–472. [Google Scholar] [CrossRef]
- Zhou, W.; Liang, H.; Zhang, X. Erythrocyte damage of crucian carp (Carassius auratus) caused by microcystin-LR: In vitro study. Fish Physiol. Biochem. 2012, 38, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Krag, A.; Bendtsen, F.; Mortensen, C.; Henriksen, J.H.; Moller, S. Effects of a single terlipressin administration on cardiac function and perfusion in cirrhosis. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.H.; Gotze, J.P.; Fuglsang, S.; Christensen, E.; Bendtsen, F.; Moller, S. Increased circulating pro-brain natriuretic peptide (proBNP) and brain natriuretic peptide (BNP) in patients with cirrhosis: Relation to cardiovascular dysfunction and severity of disease. Gut 2003, 52, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Haapamaki, J.; Roine, R.P.; Turunen, U.; Farkkila, M.A.; Arkkila, P.E. Increased risk for coronary heart disease, asthma, and connective tissue diseases in inflammatory bowel disease. J. Crohns Colitis 2011, 5, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Chen, G.; Cai, D.; Zhao, S.; Cheng, J.; Shen, H. Inflammatory Bowel Disease and Risk of Ischemic Heart Disease: An Updated Meta-Analysis of Cohort Studies. J. Am. Heart Assoc. 2017, 6, e005892. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Benedetto, F.A.; Mallamaci, F.; Tripepi, G.; Giacone, G.; Cataliotti, A.; Seminara, G.; Stancanelli, B.; Malatino, L.S. Prognostic value of echocardiographic indicators of left ventricular systolic function in asymptomatic dialysis patients. J. Am. Soc. Nephrol. 2004, 15, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Dubin, R.F.; Deo, R.; Bansal, N.; Anderson, A.H.; Yang, P.; Go, A.S.; Keane, M.; Townsend, R.; Porter, A.; Budoff, M. Associations of Conventional Echocardiographic Measures with Incident Heart Failure and Mortality: The Chronic Renal Insufficiency Cohort. Clin. J. Am. Soc. Nephrol. 2017, 12, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.P.; Chandra, A.; Navaneethan, S.D. Blood pressure parameters and morbid and mortal outcomes in nondialysis-dependent chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 16–22. [Google Scholar] [CrossRef]
- Hassan, M.O.; Duarte, R.; Dix-Peek, T.; Vachiat, A.; Naidoo, S.; Dickens, C.; Grinter, S.; Manga, P.; Naicker, S. Correlation between volume overload, chronic inflammation, and left ventricular dysfunction in chronic kidney disease patients. Clin. Nephrol. 2016, 86, 131–135. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kimura, T.; Kuragano, T. The Hepcidin-Anemia Axis: Pathogenesis of Anemia in Chronic Kidney Disease. Contrib. Nephrol. 2019, 198, 124–134. [Google Scholar]
- Steiner, K.; Zimmermann, L.; Hagenbuch, B.; Dietrich, D. Zebrafish Oatp-mediated transport of microcystin congeners. Arch. Toxicol. 2016, 90, 1129–1139. [Google Scholar] [CrossRef]
- Hausner, E.A.; Elmore, S.A.; Yang, X. Overview of the Components of Cardiac Metabolism. Drug Metab. Dispos. 2019, 47, 673–688. [Google Scholar] [CrossRef]
- Fujiwara, K.; Adachi, H.; Nishio, T.; Unno, M.; Tokui, T.; Okabe, M.; Onogawa, T.; Suzuki, T.; Asano, N.; Tanemoto, M. Identification of thyroid hormone transporters in humans: Different molecules are involved in a tissue-specific manner. Endocrinology 2001, 142, 2005–2012. [Google Scholar] [CrossRef]
- Lu, R.; Kanai, N.; Bao, Y.; Schuster, V.L. Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT). J. Clin. Investig. 1996, 98, 1142–1149. [Google Scholar] [CrossRef]
- Grube, M.; Kock, K.; Oswald, S.; Draber, K.; Meissner, K.; Eckel, L.; Bohm, M.; Felix, S.B.; Vogelgesang, S.; Jedlitschky, G. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006, 80, 607–620. [Google Scholar] [CrossRef]
- Huber, R.D.; Gao, B.; Sidler Pfandler, M.A.; Zhang-Fu, W.; Leuthold, S.; Hagenbuch, B.; Folkers, G.; Meier, P.J.; Stieger, B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am. J. Physiol. Cell Physiol. 2007, 292, C795–C806. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, P.; Chen, J.; Liang, G. Distribution of microcystins in various organs (heart, liver, intestine, gonad, brain, kidney and lung) of Wistar rat via intravenous injection. Toxicon 2008, 52, 721–727. [Google Scholar] [CrossRef]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef]
- Varga, I.; Kyselovic, J.; Galfiova, P.; Danisovic, L. The Non-cardiomyocyte Cells of the Heart. Their Possible Roles in Exercise-Induced Cardiac Regeneration and Remodeling. Adv. Exp. Med. Biol. 2017, 999, 117–136. [Google Scholar]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Singal, P.K.; Iliskovic, N.; Li, T.; Kumar, D. Adriamycin cardiomyopathy: Pathophysiology and prevention. FASEB J. 1997, 11, 931–936. [Google Scholar] [CrossRef]
- Xu, M.F.; Tang, P.L.; Qian, Z.M.; Ashraf, M. Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sci. 2001, 68, 889–901. [Google Scholar] [CrossRef]
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef]
- Moreno, I.M.; Mate, A.; Repetto, G.; Vazquez, C.M.; Camean, A.M. Influence of microcystin-LR on the activity of membrane enzymes in rat intestinal mucosa. J. Physiol. Biochem. 2003, 59, 293–299. [Google Scholar] [CrossRef]
- Moreno, I.; Pichardo, S.; Jos, A.; Gomez-Amores, L.; Mate, A.; Vazquez, C.M.; Camean, A.M. Antioxidant enzyme activity and lipid peroxidation in liver and kidney of rats exposed to microcystin-LR administered intraperitoneally. Toxicon 2005, 45, 395–402. [Google Scholar] [CrossRef]
- Botha, N.; van de Venter, M.; Downing, T.G.; Shephard, E.G.; Gehringer, M.M. The effect of intraperitoneally administered microcystin-LR on the gastrointestinal tract of Balb/c mice. Toxicon 2004, 43, 251–254. [Google Scholar] [CrossRef]
- Ding, W.X.; Shen, H.M.; Ong, C.N. Critical role of reactive oxygen species formation in microcystin-induced cytoskeleton disruption in primary cultured hepatocytes. J. Toxicol. Environ. Health A 2001, 64, 507–519. [Google Scholar] [CrossRef]
- Ding, W.X.; Nam Ong, C. Role of oxidative stress and mitochondrial changes in cyanobacteria-induced apoptosis and hepatotoxicity. FEMS Microbiol. Lett. 2003, 220, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef]
- Choy, K.W.; Murugan, D.; Mustafa, M.R. Natural products targeting ER stress pathway for the treatment of cardiovascular diseases. Pharmacol. Res. 2018, 132, 119–129. [Google Scholar] [CrossRef]
- Glembotski, C.C. Endoplasmic reticulum stress in the heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell. Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef]
- Kim, A.J.; Shi, Y.; Austin, R.C.; Werstuck, G.H. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J. Cell Sci. 2005, 118, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Takada, A.; Miki, T.; Kuno, A.; Kouzu, H.; Sunaga, D.; Itoh, T.; Tanno, M.; Yano, T.; Sato, T.; Ishikawa, S. Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. PLoS ONE 2012, 7, e39893. [Google Scholar] [CrossRef]
- Kassan, M.; Galan, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef]
- Young, C.N.; Cao, X.; Guruju, M.R.; Pierce, J.P.; Morgan, D.A.; Wang, G.; Iadecola, C.; Mark, A.L.; Davisson, R.L. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J. Clin. Investig. 2012, 122, 3960–3964. [Google Scholar] [CrossRef]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef]
- Ferrandi, C.; Ballerio, R.; Gaillard, P.; Giachetti, C.; Carboni, S.; Vitte, P.A.; Gotteland, J.P.; Cirillo, R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004, 142, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Liu, J.; Li, C.; Wang, J. Critical Role of Endoplasmic Reticulum Stress in Cognitive Impairment Induced by Microcystin-LR. Int. J. Mol. Sci. 2015, 16, 28077–28086. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Liu, J.; Li, C.; Wang, J. Intracellular Calcium Plays a Critical Role in the Microcystin-LR-Elicited Neurotoxicity through PLC/IP3 Pathway. Int. J. Toxicol. 2015, 34, 551–558. [Google Scholar] [CrossRef]
- Zhao, S.; Li, G.; Chen, J. A proteomic analysis of prenatal transfer of microcystin-LR induced neurotoxicity in rat offspring. J. Proteom. 2015, 114, 197–213. [Google Scholar] [CrossRef]
- Zhou, M.; Tu, W.W.; Xu, J. Mechanisms of microcystin-LR-induced cytoskeletal disruption in animal cells. Toxicon 2015, 101, 92–100. [Google Scholar] [CrossRef]
- Lubbers, E.R.; Mohler, P.J. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J. Mol. Cell. Cardiol. 2016, 101, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Ginsburg, K.S.; Hall, D.D.; Zimmermann, M.; Stein, I.S.; Zhang, M.; Tandan, S.; Hill, J.A.; Horne, M.C.; Bers, D. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Ca v1.2. Biochemistry 2010, 49, 10298–10307. [Google Scholar] [CrossRef]
- Bhasin, N.; Cunha, S.R.; Mudannayake, M.; Gigena, M.S.; Rogers, T.B.; Mohler, P.J. Molecular basis for PP2A regulatory subunit ubunit B56alpha targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H109–H119. [Google Scholar] [CrossRef]
- Meyer-Roxlau, S.; Lammle, S.; Opitz, A.; Kunzel, S.; Joos, J.P.; Neef, S.; Sekeres, K.; Sossalla, S.; Schondube, F.; Alexiou, K. Differential regulation of protein phosphatase 1 (PP1) isoforms in human heart failure and atrial fibrillation. Basic Res. Cardiol. 2017, 112, 43. [Google Scholar] [CrossRef]
- Fontanillo, M.; Kohn, M. Microcystins: Synthesis and structure-activity relationship studies toward PP1 and PP2A. Bioorg. Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef]
- Johnson, L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009, 37, 627–641. [Google Scholar] [CrossRef]
- Heijman, J.; Ghezelbash, S.; Wehrens, X.H.; Dobrev, D. Serine/Threonine Phosphatases in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2017, 103, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.D.; Feekes, J.A.; Arachchige Don, A.S.; Shi, M.; Hamid, J.; Chen, L.; Strack, S.; Zamponi, G.W.; Horne, M.C.; Hell, J.W. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 2006, 45, 3448–3459. [Google Scholar] [CrossRef]
- Kimura, T.; Han, W.; Pagel, P.; Nairn, A.C.; Caplan, M.J. Protein phosphatase 2A interacts with the Na,K-ATPase and modulates its trafficking by inhibition of its association with arrestin. PLoS ONE 2011, 6, e29269. [Google Scholar] [CrossRef]
- Lecuona, E.; Dada, L.A.; Sun, H.; Butti, M.L.; Zhou, G.; Chew, T.L.; Sznajder, J.I. Na,K-ATPase alpha1-subunit dephosphorylation by protein phosphatase 2A is necessary for its recruitment to the plasma membrane. FASEB J. 2006, 20, 2618–2620. [Google Scholar] [CrossRef]
- DeGrande, S.T.; Little, S.C.; Nixon, D.J.; Wright, P.; Snyder, J.; Dun, W.; Murphy, N.; Kilic, A.; Higgins, R.; Binkley, P.F. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J. Biol. Chem. 2013, 288, 1032–1046. [Google Scholar] [CrossRef]
- Gergs, U.; Boknik, P.; Buchwalow, I.; Fabritz, L.; Matus, M.; Justus, I.; Hanske, G.; Schmitz, W.; Neumann, J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 2004, 279, 40827–40834. [Google Scholar] [CrossRef]
- Hoehn, M.; Zhang, Y.; Xu, J.; Gergs, U.; Boknik, P.; Werdan, K.; Neumann, J.; Ebelt, H. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of beta-catenin by glycogen synthase kinase 3beta. Int. J. Cardiol. 2015, 183, 39–46. [Google Scholar] [CrossRef]
- Baskaran, R.; Velmurugan, B.K. Protein phosphatase 2A as therapeutic targets in various disease models. Life Sci. 2018, 210, 40–46. [Google Scholar] [CrossRef]
- Weber, S.; Meyer-Roxlau, S.; Wagner, M.; Dobrev, D.; El-Armouche, A. Counteracting Protein Kinase Activity in the Heart: The Multiple Roles of Protein Phosphatases. Front. Pharmacol. 2015, 6, 270. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Zhong, T.P. Zebrafish genetics and formation of embryonic vasculature. Curr. Top. Dev. Biol. 2005, 71, 53–81. [Google Scholar]
- Curry, F.R.; Adamson, R.H. Vascular permeability modulation at the cell, microvessel, or whole organ level: Towards closing gaps in our knowledge. Cardiovasc. Res. 2010, 87, 218–229. [Google Scholar] [CrossRef]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Sturtzel, C. Endothelial Cells. In The Immunology of Cardiovascular Homeostasis and Pathology; Springer: Berlin, Germany, 2017; pp. 71–91. [Google Scholar]
- Komatsu, M.; Furukawa, T.; Ikeda, R.; Takumi, S.; Nong, Q.; Aoyama, K.; Akiyama, S.; Keppler, D.; Takeuchi, T. Involvement of mitogen-activated protein kinase signaling pathways in microcystin-LR-induced apoptosis after its selective uptake mediated by OATP1B1 and OATP1B3. Toxicol. Sci. 2007, 97, 407–416. [Google Scholar] [CrossRef]
- Falconer, I.R.; Yeung, D.S. Cytoskeletal changes in hepatocytes induced by microcystis toxins and their relation to hyperphosphorilation of cell proteins. Chem. Biol. Interact. 1992, 81, 181–196. [Google Scholar] [CrossRef]
- Strack, S.; Cribbs, J.T.; Gomez, L. Critical role for protein phosphatase 2A heterotrimers in mammalian cell survival. J. Biol. Chem. 2004, 279, 47732–47739. [Google Scholar] [CrossRef]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef]
- Weng, D.; Lu, Y.; Wei, Y.; Liu, Y.; Shen, P. The role of ROS in microcystin-LR-induced hepatocyte apoptosis and liver injury in mice. Toxicology 2007, 232, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.M.; Govender, S.; Shah, M.; Downing, T.G. An investigation of the role of vitamin E in the protection of mice against microcystin toxicity. Environ. Toxicol. 2003, 18, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.; Anand, T.; Rao, P.V. Activity and gene expression profile of certain antioxidant enzymes to microcystin-LR induced oxidative stress in mice. Toxicology 2006, 220, 136–146. [Google Scholar] [CrossRef]
- Gehringer, M.M.; Shephard, E.G.; Downing, T.G.; Wiegand, C.; Neilan, B.A. An investigation into the detoxification of microcystin-LR by the glutathione pathway in Balb/c mice. Int. J. Biochem. Cell Biol. 2004, 36, 931–941. [Google Scholar] [CrossRef]
- Mellinger, J.L.; Pencina, K.M.; Massaro, J.M.; Hoffmann, U.; Seshadri, S.; Fox, C.S.; O’Donnell, C.J.; Speliotes, E.K. Hepatic steatosis and cardiovascular disease outcomes: An analysis of the Framingham Heart Study. J. Hepatol. 2015, 63, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sookoian, S.; Pirola, C.J. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J. Hepatol. 2008, 49, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Oni, E.T.; Agatston, A.S.; Blaha, M.J. A systematic review: Burden and severity of subclinical cardiovascular disease among those with nonalcoholic fatty liver; should we care? Atherosclerosis 2013, 230, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, S.; Perseghin, G.; Molon, G. Nonalcoholic fatty liver disease is associated with left ventricular diastolic dysfunction in patients with type 2 diabetes. Diabetes Care 2012, 35, 389–395. [Google Scholar] [CrossRef] [PubMed]
- VanWagner, L.B.; Wilcox, J.E.; Colangelo, L.A. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: A population-based study. Hepatology 2015, 62, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Hallsworth, K.; Hollingsworth, K.G.; Thoma, C. Cardiac structure and function are altered in adults with non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 757–762. [Google Scholar] [CrossRef]
- Boddi, M.; Tarquini, R.; Chiostri, M. Nonalcoholic fatty liver in nondiabetic patients with acute coronary syndromes. Eur. J. Clin. Investig. 2013, 43, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.K.; Honar, H.; Liu, H.; ter Keurs, H.E.; Lee, S.S. Role of cardiac myofilament proteins titin and collagen in the pathogenesis of diastolic dysfunction in cirrhotic rats. J. Hepatol. 2011, 55, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, A.M.; Barents, M.; de Jongste, M.J.; Romer, J.W.; Steward, R.N.; Muskiet, F.A. High Intraindividual Variation of N-Terminal Pro-B-Type Natriuretic Peptide in Urine of Patients with Stable Chronic Heart Failure: Comparison with Plasma. Clin. Chem. 2016, 62, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zheng, X.L. Correlation of serum brain natriuretic peptide level, severity and cardiac function of liver disease in patients with hepatitis C viral cirrhosis and nonalcoholic fatty liver. Modern Med. 2016, 44, 1675–1680. [Google Scholar]
- Kao, M.P.; Ang, D.S.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, J.; Feng, Y.; Zhang, J.; Cui, L.; Zhang, H.; Bai, Y. Extracellular acidosis suppresses calcification of vascular smooth muscle cells by inhibiting calcium influx via L-type calcium channels. Clin. Exp. Hypertens. 2018, 40, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Falconer, I.R.; Dornbusch, M.; Moran, G.; Yeung, S.K. Effect of the cyanobacterial (blue-green algal) toxins from Microcystis aeruginosa on isolated enterocytes from the chicken small intestine. Toxicon 1992, 30, 790–793. [Google Scholar] [CrossRef]
- Botha, N.; Gehringer, M.M.; Downing, T.G.; van de Venter, M.; Shephard, E.G. The role of microcystin-LR in the induction of apoptosis and oxidative stress in CaCo2 cells. Toxicon 2004, 43, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, X.; Yu, B.; Yu, G. Characterization of in vitro effects of microcystin-LR on intestinal epithelial cells. Environ. Toxicol. 2017, 32, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, J.; Huet, S.; Jarry, G.; Fessard, V. In vivo DNA damage induced by the cyanotoxin microcystin-LR: Comparison of intra-peritoneal and oral administrations by use of the comet assay. Mutat. Res. 2008, 652, 65–71. [Google Scholar] [CrossRef]
- Nobre, A.C.L.; Nunes-Monteiro, S.M.; Monteiro, M.C.; Martins, A.M.; Havt, A.; Barbosa, P.S.; Lima, A.A.; Monteiro, H.S. Microcystin-LR promote intestinal secretion of water and electrolytes in rats. Toxicon 2004, 44, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Huguet, A.; Henri, J.; Petitpas, M.; Hogeveen, K.; Fessard, V. Comparative cytotoxicity, oxidative stress, and cytokine secretion induced by two cyanotoxin variants, microcystin LR and RR, in human intestinal Caco-2 cells. J. Biochem. Mol. Toxicol. 2013, 27, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Puerto, M.; Pichardo, S.; Jos, A.; Camean, A.M. Microcystin-LR induces toxic effects in differentiated and undifferentiated Caco-2 cells. Arch. Toxicol. 2010, 84, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, C.; Zhang, T.; Liu, W.; Wang, L.; Chen, Y.; Wu, L.; Hegazy, A.M.; El-Sayed, A.F.; Zhang, X. muEvaluation of microcystin-LR absorption using an in vivo intestine model and its effect on zebrafish intestine. Aquat. Toxicol. 2019, 206, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Kondo, F.; Harada, K. Hepatic necrosis in aged mice by oral administration of microcystin-LR. Toxicon 1997, 35, 231–239. [Google Scholar] [CrossRef]
- Lin, J.; Chen, J.; He, J.; Chen, J.; Yan, Q.; Zhou, J.; Xie, P. Effects of microcystin-LR on bacterial and fungal functional genes profile in rat gut. Toxicon 2015, 96, 50–56. [Google Scholar] [CrossRef]
- Chen, J.; Xie, P.; Lin, J.; He, J.; Zeng, C.; Chen, J. Effects of microcystin-LR on gut microflora in different gut regions of mice. J. Toxicol. Sci. 2015, 40, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Anker, S.D.; Egerer, K.R.; Volk, H.D.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am. J. Cardiol. 1997, 79, 1426–1430. [Google Scholar] [CrossRef]
- Sandek, A.; Bauditz, J.; Swidsinski, A.; Buhner, S.; Weber-Eibel, J.; von Haehling, S.; Schroedl, W.; Karhausen, T.; Doehner, W.; Rauchhaus, M. Altered intestinal function in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 50, 1561–1569. [Google Scholar] [CrossRef]
- Zhao, L.H.; Lu, C.L.; Sun, H.T. New progress in the study of the relationship between intestinal flora and mucous membrane immunity and cardiovascular disease. Chin. J. Immunol. 2018, 34, 1433–1436. [Google Scholar]
- Long, M.T.; Ko, D.; Arnold, L.M.; Trinquart, L.; Sherer, J.A.; Keppel, S.S.; Benjamin, E.J.; Helm, R.H. Gastrointestinal and liver diseases and atrial fibrillation: A review of the literature. Ther. Adv. Gastroenterol. 2019, 12, 1756284819832237. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.A.; Pace, J.G.; Matson, C.F.; Miura, G.A.; Lawrence, W.B. Tissue distribution, excretion and hepatic biotransformation of microcystin-LR in mice. J. Pharmacol. Exp. Ther. 1991, 256, 176–182. [Google Scholar] [PubMed]
- Hagenbuch, B.; Meier, P.J. The superfamily of organic anion transporting polypeptides. Biochim. Biophys. Acta 2003, 1609, 1–18. [Google Scholar] [CrossRef]
- Menezes, C.; Valerio, E.; Dias, E. The Kidney Bero-E6 Cell Line: A Suitable Model to Study the Toxicity of Microcystins. In New Insights into Toxicity and Drug Testing; IntechOpen: London, UK, 2013; pp. 29–48. [Google Scholar]
- Jia, J.; Luo, W.; Lu, Y.; Giesy, J.P. Bioaccumulation of microcystins (MCs) in four fish species from Lake Taihu, China: Assessment of risks to humans. Sci. Total Environ. 2014, 487, 224–232. [Google Scholar] [CrossRef]
- Wang, Z.; Li, G.; Wu, Q.; Liu, C.; Shen, J.; Yan, W. Microcystin-LR exposure induced nephrotoxicity by triggering apoptosis in female zebrafish. Chemosphere 2019, 214, 598–605. [Google Scholar] [CrossRef]
- McLellan, N.L.; Manderville, R.A. Toxic mechanisms of microcystins in mammals. Toxicol. Res. 2017, 6, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, E.; Matos, P.; Pereira, P.; Batoreu, M.C.; Silva, M.J.; Jordan, P. Microcystin-LR activates the ERK1/2 kinases and stimulates the proliferation of the monkey kidney-derived cell line Vero-E6. Toxicol. In Vitro 2010, 24, 1689–1695. [Google Scholar] [CrossRef]
Figure 1.
General chemical structure of MCs (microcystins). (A) 1–7 represent seven amino acid residues, respectively. X and Z in positions two and four are highly variable L-amino acids that determine the suffix in the nomenclature of MCs. (B) represents some of the most frequent MC congeners (adapted from Chen et al. [19]).
Figure 1.
General chemical structure of MCs (microcystins). (A) 1–7 represent seven amino acid residues, respectively. X and Z in positions two and four are highly variable L-amino acids that determine the suffix in the nomenclature of MCs. (B) represents some of the most frequent MC congeners (adapted from Chen et al. [19]).

Figure 2.
Possible mechanisms of indirect cardiovascular toxicity induced by MCs. MCs are capable of causing cardiovascular disease by inducing pathological changes in structure and/or function of liver, gastrointestinal tract and kidney, namely indirect cardiovascular toxicity of MCs. Abbreviation: RAS: renin-angiotensin system; ROS: reactive oxygen species.
Figure 2.
Possible mechanisms of indirect cardiovascular toxicity induced by MCs. MCs are capable of causing cardiovascular disease by inducing pathological changes in structure and/or function of liver, gastrointestinal tract and kidney, namely indirect cardiovascular toxicity of MCs. Abbreviation: RAS: renin-angiotensin system; ROS: reactive oxygen species.

Figure 3.
Possible mechanisms of cardiovascular toxicity caused by MCs. MCs are transported into cells through OATP (organic anion transporting polypeptides). Having entered the cells, MCs can cause alterations in expression level of some genes, increase the level of ROS, induce oxidative stress in mitochondria and ER, change the modification of related proteins (such as phosphorylation and dephosphorization of proteins) and destroy the cytoskeleton, subsequently playing direct or indirect toxicity to the cardiovascular system, and finally inducing the occurrence of CVD.
Figure 3.
Possible mechanisms of cardiovascular toxicity caused by MCs. MCs are transported into cells through OATP (organic anion transporting polypeptides). Having entered the cells, MCs can cause alterations in expression level of some genes, increase the level of ROS, induce oxidative stress in mitochondria and ER, change the modification of related proteins (such as phosphorylation and dephosphorization of proteins) and destroy the cytoskeleton, subsequently playing direct or indirect toxicity to the cardiovascular system, and finally inducing the occurrence of CVD.


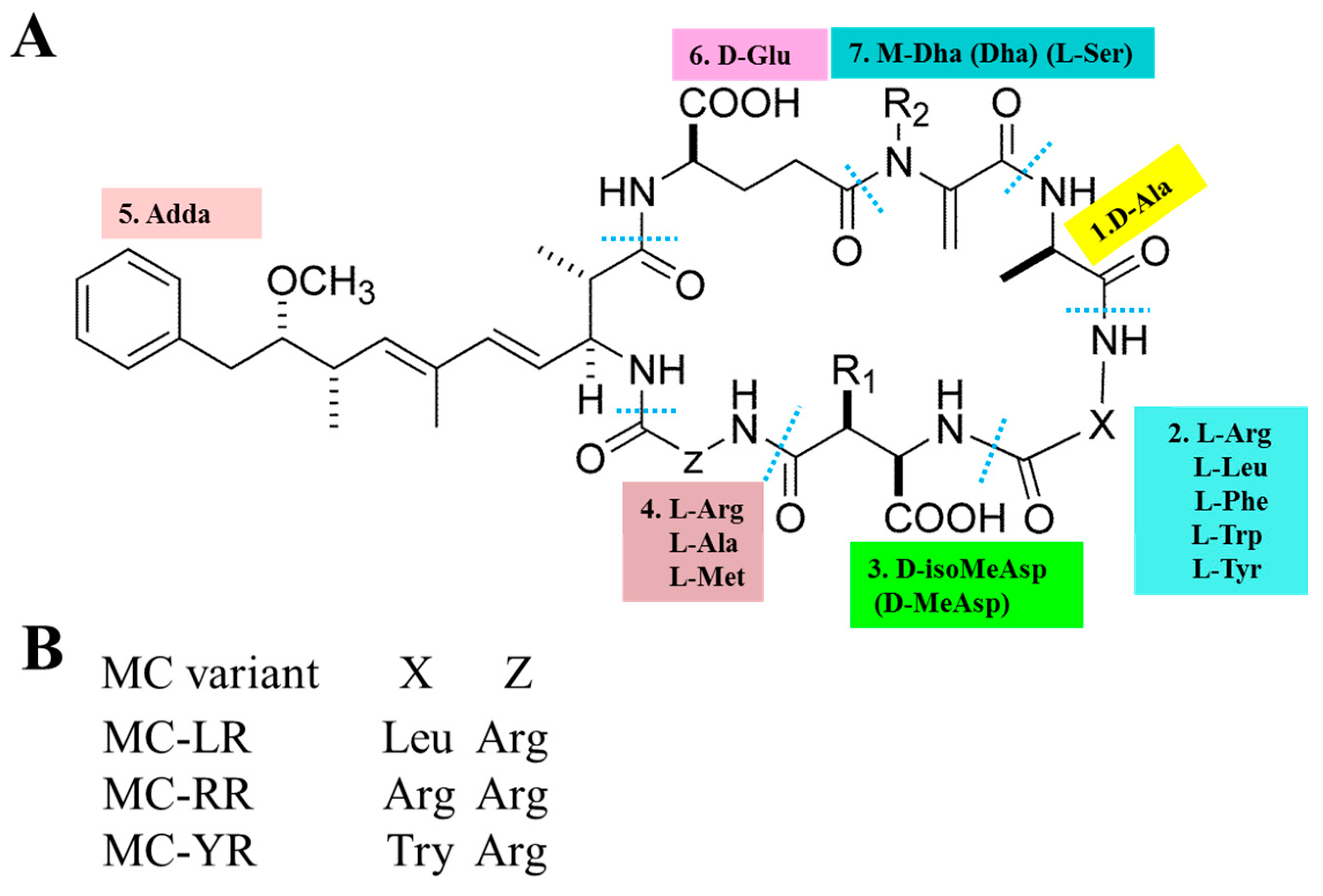{kind=link}
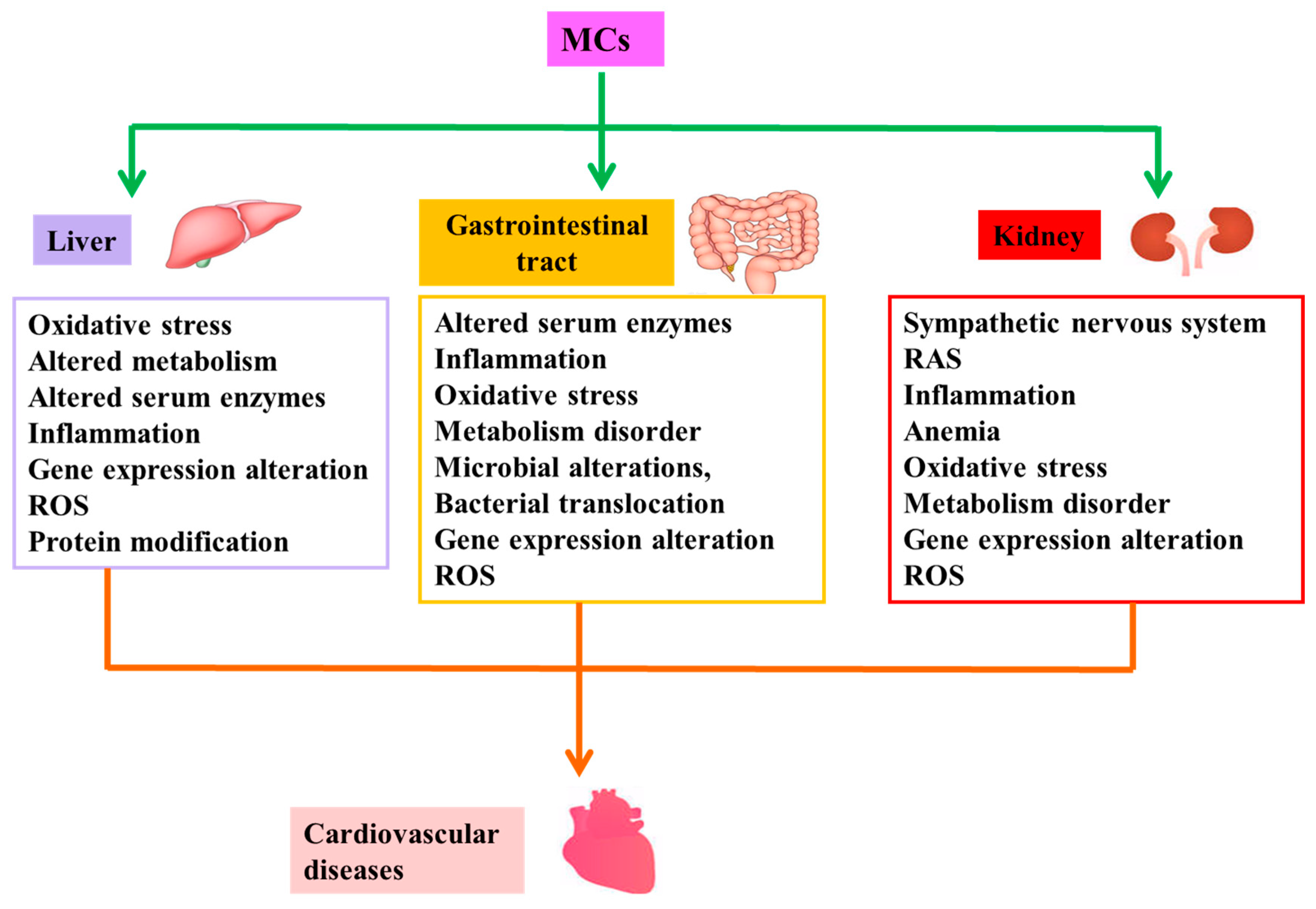{kind=link}
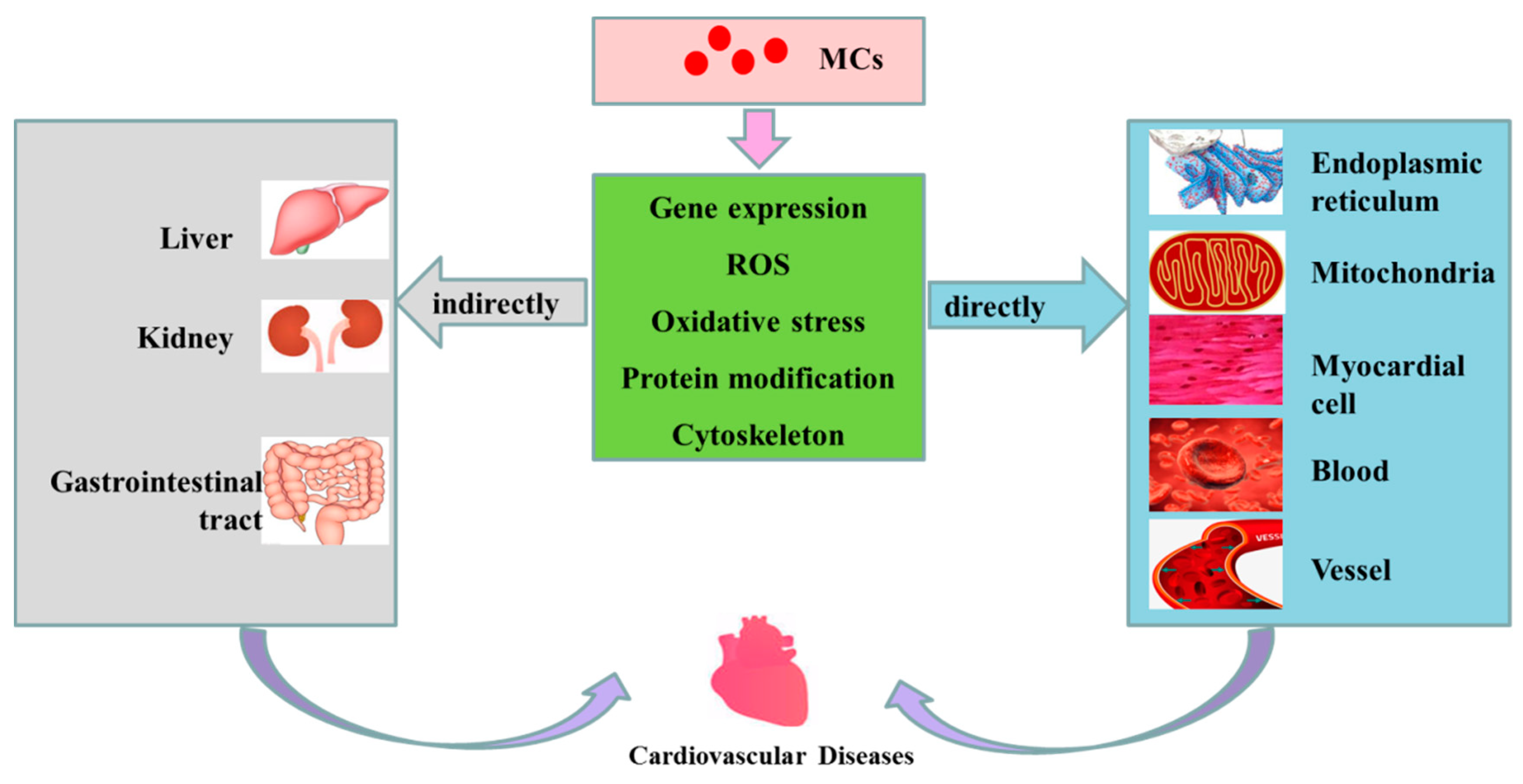{kind=link}
Table 1.
Summary of cardiovascular toxicity of microcystins in vivo studies.
| Animals | Exposure | Toxicant | Dose | Time Point | Toxic Effects | References |
|---|---|---|---|---|---|---|
| Rabbits | I.P | MCs (mainly containing MC-RR, MC-LR | 12.5, 50 μg/kg | 1, 3, 12, 24, 48 h | Damage of mitochondrial morphology, lipid peroxidation ↑, SDH ↑, Ca2+-Mg2+-ATPase activities of mitochondria ↓, disrupted ionic homeostasis, MMP ↓ | [36] |
| SD rats | I.P | MC-LR | 122 μg/kg | 1, 7, 14 day | Myocardial cells damage including pyknosis, plasma dissolve and myofibrilla necrosis, LDH ↑, AST ↑, CK ↑ | [37] |
| loach, Misguruns mizolepis Gunthe embryos | orally | MC-LR | 1, 3, 10, 100, 1000 μg/L | 7 days | Pericardial edema, tubular heart and bradycardia | [38] |
| Wistar rats | I.P | MC-LR | 10 μg/kg | every two days for 8 months | Destruction cytoskeletons of cardiomyocytes, loss of cell crossstriations, myofibril volume fraction ↓, cardiomyocytes volume ↑,myofibrillar volume ↓, even fibrosis, infiltration of mononuclear in the interstitial tissue | [39] |
| Male Wistar rats | I.P | MC-YR | 10 μg/kg | every two days for 8 months | Volume density of cardiac muscle tissue ↓, fibrous proliferation, lymphocyte infiltration, enlarged and often bizarre-shaped nuclei, myofibril volume fraction ↓ | [40] |
| Male Wistar rats | I.P | MC-LR | 0.16 LD50 (14 g/kg), 1 LD50 (87 g/kg) | 24 h | Myocardial infarction in dead rats, CK ↑, troponin I ↑, GSH ↑, lipid peroxides ↑, antioxidant enzymes ↑, heart rate ↓, blood pressure ↓, loss of adhesion between cardiomyocytes, swelling or rupture of mitochondria, complex I/III ↓, electron flow along the mitochondrial respiratory chain ↓ | [41] |
| Zebrafish | orally | MC-LR | 0.1, 1 μM | 24 h | Angiodysplasia, damaged vascular structures, lumen size ↓, blood flow ↓, vascular dysfunction | [42] |
| Trahira, hoplias malabaricus | orally | MC-LR | 100 μg/kg | 48 h | GPx ↑, GSH ↓ | [43] |
| Japanese medaka (Oryzias latipes) | orally | MC-LR | 600, 6300 μg/L | 15 days | Heart rate ↓, bradycardia | [44] |
| zebrafish juvenile fish | orally | MC-LR | 4.0 mM | 96h | Heart rate ↓, apoptosis ↑, morphological deformity, stunted growth | [45] |
| Male Fischer 344 Rats | orally | MC-LR | 100 μg/kg | Cardiac output and stroke volume ↓, an acute hypotension responsive to volume, expansion with whole blood; heart rate ↓, oxygen consumption ↓, carbon dioxide production ↓, metabolic rate ↓, progressive hypothermia, acid-base balance changes | [46] | |
| KM (Kunming) male mice | I.P | MC-LR | 0, 3.125, 6.25, 12.5, 25 μg/kg/day | 7 days | ALP ↑, LDH ↑, AST ↑, ALT ↑, RBC↓, HGB↓, HCT ↓, MCV -, MCH -, MCHC -, WBC ↑, Mon ↑, Gran ↑ | [47] |
| Male Wistar albino rats | orally | MCs (mainly containing MC-LR, MC-YR, MC-RR, MC-LF, MC-LW) | 25,000 μg/kg of food | 28 days | RBC ↓, MCV ↑, MCH ↓, MCHC ↑ | [48] |
Table 2.
Summary of cardiovascular toxicity of microcystins in vitro studies.
| Cells | Toxicant | Dose | Time Point | Toxic Effects | References |
|---|---|---|---|---|---|
| H9C2 (rat cardiomyocytes) | MC-LR | 10 μM | 0, 4, 8, 12, 16, 20, 24 h | Expression levels of rhythmic genes ↓, antioxidant genes ↑ | [49] |
| Human umbilical vein endothelial cells (HUVECs) | MC-LR | 0.1, 1 μM | 24 h | Apoptosis ↑, caspase3/9 ↑, mitochondrial ROS ↑, MMP ↓, p53 ↑, PCNA ↓ | [42] |
| HUVECs | MC-LR | 40 μM | 24 h | Proliferation ↓, apoptosis ↑, migration ↓, capillary-like structure formation ↓, ROS ↑, NF-κB ↑, TNF-α ↑, VCAM-1 ↑, ICAM-1 ↑ | [50] |
| HUVECs | MC-LR | 40 μM | 24 h | Cell death ↑, cell viability ↓, migration ↓, tube formation ↓, ROS ↑, NF-κB ↑, TNF-α ↑, IL6 ↑, SOD ↓, GSH ↓, MDA ↑ | [51] |
| HUVECs | MC-LR | 40 μM | 24 h | MMP ↓, decreased levels of cytochrome c ↓, caspase-3/-9 ↑, ROS ↑, apoptosis ↑, NRF 2/ HO 1 ↓ | [52] |
| Crucian carp erythrocytes | MC-LR | 0, 1, 10, 100, 1000 nM. | 1, 3, 12, 24, 48 h | GSH ↑, SOD ↑, CAT ↑, GPx ↑, GST ↑, GLT ↓, lipid peroxidate ↑, hemolysis ↑, AchE ↓, Ca2+-Mg2+-ATPase ↓, Na2+-k+-ATPase ↓ | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cao, L.; Massey, I.Y.; Feng, H.; Yang, F. A Review of Cardiovascular Toxicity of Microcystins. Toxins 2019, 11, 507. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090507
AMA Style
Cao L, Massey IY, Feng H, Yang F. A Review of Cardiovascular Toxicity of Microcystins. Toxins. 2019; 11(9):507. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090507
Chicago/Turabian StyleCao, Linghui, Isaac Yaw Massey, Hai Feng, and Fei Yang. 2019. "A Review of Cardiovascular Toxicity of Microcystins" Toxins 11, no. 9: 507. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090507
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.